Pulmonary hypertension means high blood pressure in the lungs. This is an extremely important issue in scleroderma occurring in up to 40% of patients.
Patients with limited scleroderma have the risk of developing progressive blood vessel narrowing in the lungs frequently in the absence of lung scarring and inflammation. This complication is called pulmonary arterial hypertension (PAH). PAH is recognizedby the World Health Organization (WHO) as a distinct medical syndrome that shares common tissue features of non-inflammatory blood vessel narrowing. PAH occurs in scleroderma but also in lupus, as an isolated disease called idiopathic PAH, and as a complication of liver failure, HIV infection and use of diet pills.
PAH can occur in diffuse scleroderma as well but a more common scenario is for progressive lung scarring to lead to loss of microvasculature in the lung again leading to elevated lung blood pressure. This syndrome is recognized by the WHO as pulmonary hypertension (PH) secondary to intrinsic lung disease.
Symptoms
The most common symptom of PAH and PH is shortness of breath with physical activity. Fainting or near fainting with physical activity may occur as well as other symptoms such as fluid retention and chest discomfort.
The diagnosis of PAH and PH are often overlooked by the community physician. It is sufficiently common and of such high impact that all patients with scleroderma should be screened for its presence on a regular basis.
Pathophysiology
Blood returning to the lungs is pumped through the lungs by the right ventricle of the heart. Blood pressure in the lungs is ordinarily rather low, for example 20/10, in contrast to body blood pressure which is usually around 110/70.
Resistance to blood flow through the lungs puts a strain on the right ventricle. At early stages, the right ventricle is able to compensate but as the resistance increases and the pressures go higher, the right ventricle cannot keep up. At early stages, shortness of breath occurs with moderate physical activity but as it worsens, it takes less and less physical activity to cause shortness of breath.
The pressure in the lungs can be elevated for several different reasons in scleroderma.
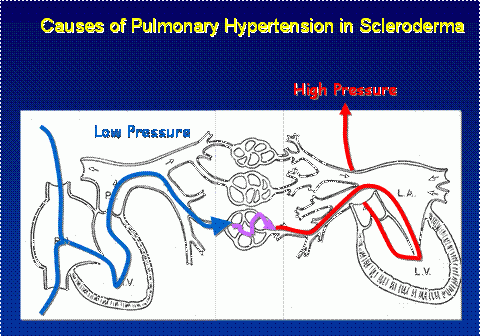
In scleroderma, the left ventricle can become stiff or weakened, leading to back pressure into the lungs (red arrow). This is called heart failure. In patients with lung scarring (see Lung Involvement), the capillaries can be damaged (lavender arrow) which leads to PH. The distinctive problem in scleroderma is narrowing of the small lung arteries (blue arrow). This is the problem that leads to PAH.
How is PAH/PH Diagnosed?
The pulmonary function tests can be helpful. Patients in whom the diffusing capacity is reduced either as an isolated finding or out of proportion to changes in their forced vital capacity are more likely to have pulmonary hypertension. Blood pressure in the lung can be estimated and the size and function of the right ventricle assessed by echocardiography with Doppler. This non-invasive test uses sound waves to image the chambers of the heart. However, Doppler echocardiograms are notoriously inaccurate when the pulmonary hypertension is mild or when there is simultaneous presence of lung scarring. A small protein released by stretched heart muscle (BNP-brain natriuretic peptide) can be measured by a simple blood test. Elevated levels are a strong clue for suspecting pulmonary hypertension.
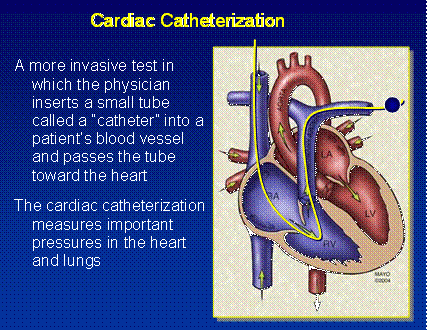
PULMONARY HYPERTENSION CANNOT BE DIAGNOSED WITHOUT RIGHT HEART CATHETERIZATION
This is an invasive test performed by specialists in cardiology wherein a slender tube is introduced into the circulation, advanced carefully to the right side of the heart and into the lungs. Careful measurements of blood flows and pressures define the diagnosis of pulmonary hypertension.
What Causes Pulmonary Hypertension?
As with many features of scleroderma, the basic issue is progressive scarring of the inner lining of the small artery. The changes in the lung blood vessels look remarkably similar to those in the fingers, kidneys and gastrointestinal tract. Many in the research community think of scleroderma as a blood vessel narrowing disease and view the immune system activation and tissue scarring as secondary events.
We do not yet understand the specific triggers of blood vessel injury or just how the blood vessel damage progresses. We do understand that injury to the lining of the blood vessels leads to specific chemical imbalances that participate in pulmonary hypertension.
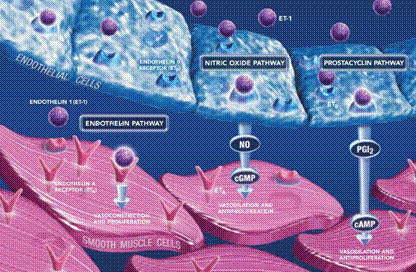
Injury to the endothelial cells which line healthy blood vessels leads to overproduction of endothelin – a key cause of blood vessel scarring and spasm – and to reduced production of nitric oxide and prostacyclins – two key body chemicals which keep blood vessels relaxed and open.
Treatment
The treatment options for PAH complicating scleroderma represent the most active area of progress in the history of the disease. The only drugs approved by the Food & Drug Administration for use in scleroderma are agents recently approved for pulmonary hypertension. These include Flolan® (an intravenous prostacyclin), Treprostinil® (a prostacyclin that is given either subcutaneously or intravenously), Ventavis® (an inhaled prostacyclin), Tracleer ® (a pill taken by mouth) and Revatio ® (a pill containing the active ingredient in Viagra).
Other similar drugs are at late stages of preapproval clinical trials and there is great interest in studying the benefits of various combination therapies.
All of these agents are uniquely and specifically suited to the blood vessel issues of PAH.
Tracleer® blocks the actions of endothelin. Revatio® accentuates the actions of nitric oxide. Flolan®, Treprostinil® and Ventavis® are artificial prostacyclins designed to replace what the damaged blood vessel no longer can produce.
These agents are very complex and choosing the correct initial treatment requires considerable expertise. The University of Michigan Scleroderma Program works closely with the Pulmonary Hypertension Program in identifying pulmonary hypertension and in choosing the correct path in treatment.
These drugs are extraordinarily expensive, ranging from $15,000-$150,000 per year. Pharmaceutical benefit issues influence choice of therapy. All of these drugs were developed under new government programs that foster drug development for rare or “orphan” diseases. In part, because the “market” is small, the cost per prescription is high.
All of these treatments reduce shortness of breath, improve exercise capacity, and slow the rate of clinical worsening.
How is Treatment Assessed?
The most important elements in following response to treatment are the level of shortness of breath and the exercise capacity. A simple test called the “six minute walk” is done in the pulmonary function laboratory. This measures how far one can walk and what happens to blood oxygen levels during exertion. It is commonly used in assessing and adjusting treatments for pulmonary hypertension.
Other tests include serial assessments of right ventricle health by echocardiogram and by BNP blood test. Repeat catheterization studies are done when clinically indicated.
For more information, contact: Pulmonary Hypertension Association
This national organization is a model for collaboration between patients, physicians and other care givers and the pharmaceutical industry. This webpage features rich information for patients and health professionals.
Click here to access a copy of PHA's recently-published patient brochure.