Childhood Acute Myeloid Leukemia/Other Myeloid Malignancies Treatment (PDQ®): Treatment - Health Professional Information [NCI]
This information is produced and provided by the National Cancer Institute (NCI). The information in this topic may have changed since it was written. For the most current information, contact the National Cancer Institute via the Internet web site at http://cancer.gov or call 1-800-4-CANCER.
General Information About Childhood Acute Myeloid Leukemia (AML)
Dramatic improvements in survival have been achieved for children and adolescents with cancer.[1] Between 1975 and 2010, childhood cancer mortality decreased by more than 50%. For acute myeloid leukemia (AML), the 5-year survival rate increased over the same time from less than 20% to 68% for children younger than 15 years and from less than 20% to 57% for adolescents aged 15 to 19 years.[1]
Characteristics of Myeloid Leukemias and Other Myeloid Malignancies in Children
Approximately 20% of childhood leukemias are of myeloid origin and they represent a spectrum of hematopoietic malignancies.[2] Most myeloid leukemias are acute, and the remainder include chronic and/or subacute myeloproliferative disorders such as chronic myelogenous leukemia and juvenile myelomonocytic leukemia. Myelodysplastic syndromes occur much less frequently in children than in adults and almost invariably represent clonal, preleukemic conditions that may evolve from congenital marrow failure syndromes such as Fanconi anemia and Shwachman-Diamond syndrome.
The general characteristics of myeloid leukemias and other myeloid malignancies are described below:
- Acute myeloid leukemia (AML). AML is defined as a clonal disorder caused by malignant transformation of a bone marrow–derived, self-renewing stem cell or progenitors, leading to accumulation of immature, nonfunctional myeloid cells. These events lead to increased accumulation in the bone marrow and other organs by these malignant myeloid cells. To be called acute, the bone marrow usually must include greater than 20% immature leukemic blasts, with some exceptions as noted in subsequent sections. For more information, see the sections on Treatment Option Overview for Childhood AML and Treatment of Childhood AML.
- Transient abnormal myelopoiesis (TAM). TAM is also termed transient myeloproliferative disorder or transient leukemia. The TAM observed in infants with Down syndrome represents a clonal expansion of myeloblasts that can be difficult to distinguish from AML. Most importantly, TAM spontaneously regresses in most cases within the first 3 months of life. TAM occurs in 4% to 10% of infants with Down syndrome.[3,4,5]
TAM blasts most commonly have megakaryoblastic differentiation characteristics and distinctive mutations involving the GATA1 gene in the presence of trisomy 21.[6,7] TAM may occur in phenotypically normal infants with genetic mosaicism in the bone marrow for trisomy 21. While TAM is generally not characterized by cytogenetic abnormalities other than trisomy 21, the presence of additional cytogenetic findings may predict an increased risk of developing subsequent AML.[8] Approximately 20% of infants with TAM of Down syndrome eventually develop AML, with most cases diagnosed within the first 3 years of life.[7,8]
Early death from TAM-related complications occurs in 10% to 20% of affected infants.[8,9] Infants with progressive organomegaly, visceral effusions, high blast count (>100,000 cells/μL) and laboratory evidence of progressive liver dysfunction are at a particularly high risk of early mortality.[8,9] For more information, see the Myeloid Proliferations Associated with Down Syndrome section.
- Myelodysplastic syndrome (MDS). MDS in children represents a heterogeneous group of disorders characterized by ineffective hematopoiesis, impaired maturation of myeloid progenitors with dysplastic morphological features, and cytopenias. Although the underlying cause of MDS in children is unclear, there is often an association with marrow failure syndromes. Most patients with MDS may have hypercellular bone marrows without increased numbers of leukemic blasts, but some patients may present with a very hypocellular bone marrow, making the distinction between severe aplastic anemia and MDS difficult.[10,11]
The presence of a karyotype abnormality in a hypocellular marrow is consistent with MDS and transformation to AML should be expected. Given the high association of MDS evolving into AML, patients with MDS are typically referred for stem cell transplantation before transformation to AML. For more information, see the Myelodysplastic Syndromes (MDS) section.
- Juvenile myelomonocytic leukemia (JMML). JMML represents the most common myeloproliferative syndrome observed in young children. JMML occurs at a median age of 1.8 years.
JMML characteristically presents with hepatosplenomegaly, lymphadenopathy, fever, and skin rash along with an elevated white blood cell (WBC) count and increased circulating monocytes.[12] In addition, patients often have an elevated hemoglobin F, hypersensitivity of the leukemic cells to granulocyte-macrophage colony-stimulating factor (GM-CSF), monosomy 7, and leukemia cell mutations in a gene involved in RAS pathway signaling (e.g., NF1, KRAS, NRAS, PTPN11, or CBL).[12,13,14] For more information, see the Juvenile Myelomonocytic Leukemia (JMML) section.
- Chronic myelogenous leukemia (CML). CML is primarily an adult disease but represents the most common of the chronic myeloproliferative disorders in childhood, accounting for approximately 10% of childhood myeloid leukemia.[2] Although CML has been reported in very young children, most patients are aged 6 years and older.
CML is a clonal panmyelopathy that involves all hematopoietic cell lineages. While the WBC count can be extremely elevated, the bone marrow does not show increased numbers of leukemic blasts during the chronic phase of this disease. CML is caused by the presence of the Philadelphia chromosome, a translocation between chromosomes 9 and 22 (i.e., t(9;22)) resulting in fusion of the BCR and ABL1 genes. For more information, see the Chronic Myelogenous Leukemia (CML) section.
Other chronic myeloproliferative syndromes, such as polycythemia vera and essential thrombocytosis, are extremely rare in children.
Conditions Associated With Myeloid Malignancies
Genetic abnormalities (cancer predisposition syndromes) are associated with the development of AML. There is a high concordance rate of AML in identical twins; however, this is not believed to be related to genetic risk, but rather to shared circulation and the inability of one twin to reject leukemic cells from the other twin during fetal development.[15,16,17] There is an estimated twofold to fourfold increased risk of developing leukemia for the fraternal twin of a pediatric leukemia patient up to about age 6 years, after which the risk is not significantly greater than that of the general population.[18,19]
The development of AML has also been associated with a variety of inherited/familial syndromes, which are recognized as a unique category within the 2016 World Health Organization (WHO) Classification of Myeloid Neoplasms and Acute Leukemia. There are also several acquired conditions that increase the risk of developing AML. These inherited and acquired conditions can induce leukemogenesis through mechanisms that include chromosomal imbalances or instabilities, defects in DNA repair, altered cytokine receptor or signal transduction pathway activation, and altered protein synthesis.[20,21]
Inherited syndromes
- Chromosomal imbalances:
- Down syndrome.
- Familial monosomy 7.
- Chromosomal instability syndromes:
- Fanconi anemia.
- Dyskeratosis congenita.
- Bloom syndrome.
- Syndromes of growth and cell survival signaling pathway defects:
- Neurofibromatosis type 1 (particularly JMML development).
- Noonan syndrome (particularly JMML development).
- Severe congenital neutropenia (Kostmann syndrome, HAX1 mutations) and cyclic neutropenia (ELANE mutations).
- Shwachman-Diamond syndrome.
- Diamond-Blackfan anemia.
- Congenital amegakaryocytic thrombocytopenia (MPL mutations).
- CBL germline syndrome (particularly in JMML).
- Li-Fraumeni syndrome (TP53 mutations).
- Inherited thrombocytopenia and platelet disorders with germline predisposition to myeloid neoplasia (RUNX1, ANKRD26, and ETV6 mutations).
- GATA2 deficiency (GATA2 mutations).
Acquired syndromes
- Severe aplastic anemia.
- Paroxysmal nocturnal hemoglobinuria.
- Amegakaryocytic thrombocytopenia.
- Acquired monosomy 7.
The 2016 WHO classification system has categorized the myeloid neoplasms with germline predisposition as follows:
- Myeloid neoplasms with germline predisposition without a pre-existing disorder or organ dysfunction.[22]
- AML with germline CEBPA mutations.
- Myeloid neoplasms with germline DDX41 mutations.
- Myeloid neoplasms with germline predisposition and pre-existing platelet disorders.[22]
- Myeloid neoplasms with germline RUNX1 mutations.
- Myeloid neoplasms with germline ANKRD26 mutations.
- Myeloid neoplasms with germline ETV6 mutations.
- Myeloid neoplasms with germline predisposition and other organ dysfunction.[22]
- Myeloid neoplasms with germline GATA2 mutations.
- Myeloid neoplasms associated with bone marrow failure syndromes (including Fanconi anemia, Diamond-Blackfan anemia, and Shwachman-Diamond syndrome).
- Myeloid neoplasms associated with telomere biology disorders (including dyskeratosis congenita).
- JMML associated with neurofibromatosis, Noonan syndrome or Noonan syndrome–like disorders (including germline CBL mutations).
- Myeloid neoplasms associated with Down syndrome.
Nonsyndromic genetic susceptibility to AML is also being studied. For example, homozygosity for a specific IKZF1 polymorphism has been associated with an increased risk of infant AML.[23]
References:
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34. Also available online. Last accessed August 11, 2022.
- Roberts I, Alford K, Hall G, et al.: GATA1-mutant clones are frequent and often unsuspected in babies with Down syndrome: identification of a population at risk of leukemia. Blood 122 (24): 3908-17, 2013.
- Zipursky A: Transient leukaemia--a benign form of leukaemia in newborn infants with trisomy 21. Br J Haematol 120 (6): 930-8, 2003.
- Gamis AS, Smith FO: Transient myeloproliferative disorder in children with Down syndrome: clarity to this enigmatic disorder. Br J Haematol 159 (3): 277-87, 2012.
- Hitzler JK, Cheung J, Li Y, et al.: GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 101 (11): 4301-4, 2003.
- Mundschau G, Gurbuxani S, Gamis AS, et al.: Mutagenesis of GATA1 is an initiating event in Down syndrome leukemogenesis. Blood 101 (11): 4298-300, 2003.
- Massey GV, Zipursky A, Chang MN, et al.: A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood 107 (12): 4606-13, 2006.
- Gamis AS, Alonzo TA, Gerbing RB, et al.: Natural history of transient myeloproliferative disorder clinically diagnosed in Down syndrome neonates: a report from the Children's Oncology Group Study A2971. Blood 118 (26): 6752-9; quiz 6996, 2011.
- Hasle H, Niemeyer CM: Advances in the prognostication and management of advanced MDS in children. Br J Haematol 154 (2): 185-95, 2011.
- Schwartz JR, Ma J, Lamprecht T, et al.: The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun 8 (1): 1557, 2017.
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.
- Loh ML: Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol 152 (6): 677-87, 2011.
- Stieglitz E, Taylor-Weiner AN, Chang TY, et al.: The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47 (11): 1326-33, 2015.
- Zuelzer WW, Cox DE: Genetic aspects of leukemia. Semin Hematol 6 (3): 228-49, 1969.
- Miller RW: Persons with exceptionally high risk of leukemia. Cancer Res 27 (12): 2420-3, 1967.
- Inskip PD, Harvey EB, Boice JD, et al.: Incidence of childhood cancer in twins. Cancer Causes Control 2 (5): 315-24, 1991.
- Kurita S, Kamei Y, Ota K: Genetic studies on familial leukemia. Cancer 34 (4): 1098-101, 1974.
- Greaves M: Pre-natal origins of childhood leukemia. Rev Clin Exp Hematol 7 (3): 233-45, 2003.
- Puumala SE, Ross JA, Aplenc R, et al.: Epidemiology of childhood acute myeloid leukemia. Pediatr Blood Cancer 60 (5): 728-33, 2013.
- West AH, Godley LA, Churpek JE: Familial myelodysplastic syndrome/acute leukemia syndromes: a review and utility for translational investigations. Ann N Y Acad Sci 1310: 111-8, 2014.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Ross JA, Linabery AM, Blommer CN, et al.: Genetic variants modify susceptibility to leukemia in infants: a Children's Oncology Group report. Pediatr Blood Cancer 60 (1): 31-4, 2013.
Classification of Pediatric Myeloid Malignancies
French-American-British (FAB) Classification System for Childhood AML
The first comprehensive morphological-histochemical classification system for acute myeloid leukemia (AML) was developed by the FAB Cooperative Group.[1,2,3,4,5] This classification system, which has been replaced by the World Health Organization (WHO) system described below, categorized AML into major subtypes primarily on the basis of morphology and immunohistochemical detection of lineage markers.
The major subtypes of AML include the following:
- M0: Acute myeloblastic leukemia without differentiation.[6,7] M0 AML, also referred to as minimally differentiated AML, does not express myeloperoxidase (MPO) at the light microscopy level but may show characteristic granules by electron microscopy. M0 AML can be defined by expression of cluster determinant (CD) markers such as CD13, CD33, and CD117 (c-KIT) in the absence of lymphoid differentiation.
- M1: Acute myeloblastic leukemia with minimal differentiation but with the expression of MPO that is detected by immunohistochemistry or flow cytometry.
- M2: Acute myeloblastic leukemia with differentiation.
- M3: Acute promyelocytic leukemia (APL) hypergranular type. For more information, see the Acute Promyelocytic Leukemia (APL) section.
- M3v: APL, microgranular variant. Cytoplasm of promyelocytes demonstrates a fine granularity, and nuclei are often folded. M3v has the same clinical, cytogenetic, and therapeutic implications as FAB M3.
- M4: Acute myelomonocytic leukemia (AMML).
- M4Eo: AMML with eosinophilia (abnormal eosinophils with dysplastic basophilic granules).
- M5: Acute monocytic leukemia (AMoL).
- M5a: AMoL without differentiation (monoblastic).
- M5b: AMoL with differentiation.
- M6: Acute erythroid leukemia (AEL).
- M6a: Erythroleukemia.
- M6b: Pure erythroid leukemia (myeloblast component not apparent).
- M6c: Presence of myeloblasts and proerythroblasts.
- M7: Acute megakaryocytic leukemia (AMKL).
Other extremely rare subtypes of AML include acute eosinophilic leukemia and acute basophilic leukemia.
The FAB classification was superseded by the WHO classification described below but remains relevant as it forms the basis of the WHO's subcategory of AML, not otherwise specified (AML, NOS).
World Health Organization (WHO) Classification System for Childhood AML
In 2001, the WHO proposed a new classification system that incorporated diagnostic cytogenetic information and that more reliably correlated with outcome. In this classification, patients with t(8;21), inv(16), t(15;17), or KMT2A (MLL) translocations, which collectively constituted nearly half of the cases of childhood AML, were classified as AML with recurrent cytogenetic abnormalities. This classification system also decreased the bone marrow percentage of leukemic blast requirement for the diagnosis of AML from 30% to 20%; an additional clarification was made so that patients with recurrent cytogenetic abnormalities did not need to meet the minimum blast requirement to be considered an AML patient.[8,9,10]
In 2008, the WHO expanded the number of cytogenetic abnormalities linked to AML classification and, for the first time, included specific gene mutations (CEBPA and NPM) in its classification system.[11] In 2016, the WHO classification underwent revisions to incorporate the expanding knowledge of leukemia biomarkers that are significantly important to the diagnosis, prognosis, and treatment of leukemia.[12] With emerging technologies aimed at genetic, epigenetic, proteomic, and immunophenotypic classification, AML classification will certainly continue to evolve and provide informative prognostic and biologic guidelines to clinicians and researchers.
2016 WHO classification of AML and related neoplasms
- AML with recurrent genetic abnormalities:
- AML with t(8;21)(q22;q22), RUNX1::RUNX1T1 gene fusion.
- AML with inv(16)(p13.1;q22) or t(16;16)(p13.1;q22), CBFB::MYH11 gene fusion.
- APL with PML::RARA gene fusion.
- AML with t(9;11)(p21.3;q23.3), MLLT3::KMT2A gene fusion.
- AML with t(6;9)(p23;q34.1), DEK::NUP214 gene fusion.
- AML with inv(3)(q21.3;q26.2) or t(3;3)(q21.3;q26.2), GATA2, MECOM.
- AML (megakaryoblastic) with t(1;22)(p13.3;q13.3), RBM15::MKL1 gene fusion.
- AML with BCR::ABL1 gene fusion (provisional entity).
- AML with mutated NPM1.
- AML with biallelic mutations of CEBPA.
- AML with mutated RUNX1 (provisional entity).
- AML with myelodysplasia-related features.
- Therapy-related myeloid neoplasms.
- AML, NOS:
- AML with minimal differentiation.
- AML without maturation.
- AML with maturation.
- Acute myelomonocytic leukemia.
- Acute monoblastic/monocytic leukemia.
- Pure erythroid leukemia.
- Acute megakaryoblastic leukemia.
- Acute basophilic leukemia.
- Acute panmyelosis with myelofibrosis.
- Myeloid sarcoma.
- Myeloid proliferations related to Down syndrome:
- Transient abnormal myelopoiesis (TAM).
- Myeloid leukemia associated with Down syndrome.
2016 WHO classification of acute leukemias of ambiguous lineage
For the group of acute leukemias that have characteristics of both AML and acute lymphoblastic leukemia (ALL), the acute leukemias of ambiguous lineage, the WHO classification system is summarized in Table 1.[13,14] The criteria for lineage assignment for a diagnosis of mixed phenotype acute leukemia (MPAL) are provided in Table 2.[12,15]
| Condition | Definition |
|---|---|
| NOS = not otherwise specified. | |
| a Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.[13]Obtained from Haematologica/the Hematology Journal websitehttp://www.haematologica.org. | |
| Acute undifferentiated leukemia | Acute leukemia that does not express any marker considered specific for either lymphoid or myeloid lineage |
| Mixed phenotype acute leukemia with t(9;22)(q34;q11.2);BCR::ABL1gene fusion | Acute leukemia meeting the diagnostic criteria for mixed phenotype acute leukemia in which the blasts also have the (9;22) translocation or theBCR::ABL1rearrangement |
| Mixed phenotype acute leukemia with t(v;11q23);KMT2A(MLL) rearranged | Acute leukemia meeting the diagnostic criteria for mixed phenotype acute leukemia in which the blasts also have a translocation involving theKMT2Agene |
| Mixed phenotype acute leukemia, B/myeloid, NOS | Acute leukemia meeting the diagnostic criteria for assignment to both B and myeloid lineage, in which the blasts lack genetic abnormalities involvingBCR::ABL1gene fusion orKMT2A |
| Mixed phenotype acute leukemia, T/myeloid, NOS | Acute leukemia meeting the diagnostic criteria for assignment to both T and myeloid lineage, in which the blasts lack genetic abnormalities involvingBCR::ABL1gene fusion orKMT2A |
| Mixed phenotype acute leukemia, B/myeloid, NOS—rare types | Acute leukemia meeting the diagnostic criteria for assignment to both B- and T-lineage |
| Other ambiguous lineage leukemias | Natural killer–cell lymphoblastic leukemia/lymphoma |
| Lineage | Criteria |
|---|---|
| a Adapted from Arber et al.[12] | |
| b Strong defined as equal to or brighter than the normal B or T cells in the sample. | |
| Myeloid Lineage | Myeloperoxidase (flow cytometry, immunohistochemistry, or cytochemistry);or monocytic differentiation (at least two of the following: nonspecific esterase cytochemistry, CD11c, CD14, CD64, lysozyme) |
| T Lineage | Strongb cytoplasmic CD3 (with antibodies to CD3 epsilon chain);or surface CD3 |
| B Lineage | Strongb CD19 with at least one of the following strongly expressed: CD79a, cytoplasmic CD22, or CD10;or weak CD19 with at least two of the following strongly expressed: CD79a, cytoplasmic CD22, or CD10 |
Leukemias of mixed phenotype may be seen in various presentations, including the following:
- Bilineal leukemias in which there are two distinct populations of cells, usually one lymphoid and one myeloid.
- Biphenotypic leukemias in which individual blast cells display features of both lymphoid and myeloid lineage.
Biphenotypic cases represent the majority of mixed phenotype leukemias.[16] B-myeloid biphenotypic leukemias lacking the TEL::AML1 fusion have a lower rate of complete remission (CR) and a significantly worse event-free survival (EFS) compared with patients with precursor B-cell ALL.[16] Some studies suggest that patients with biphenotypic leukemia may fare better with a lymphoid, as opposed to a myeloid, treatment regimen.[17,18,19,20]; [21][Level of evidence C1] A large retrospective study from the international Berlin-Frankfurt-Münster (BFM) group demonstrated that initial therapy with an ALL-type regimen was associated with a superior outcome compared with AML-type or combined ALL/AML regimens, particularly in cases with CD19 positivity or other lymphoid antigen expression. In this study, hematopoietic stem cell transplantation (HSCT) in first CR was not beneficial, with the possible exception of cases with morphological evidence of persistent marrow disease (≥5% blasts) after the first month of treatment.[20]
WHO Classification of Bone Marrow and Peripheral Blood Findings for Myelodysplastic Syndromes
The FAB classification of myelodysplastic syndromes (MDS) was not completely applicable to children.[22,23] Traditionally, MDS classification systems have been divided into several distinct categories on the basis of the presence of the following:[23,24,25,26]
- Myelodysplasia.
- Types of cytopenia.
- Specific chromosomal abnormalities.
- Percentage of myeloblasts.
A modified classification schema for MDS and myeloproliferative disorders (MPDs) was published by the WHO in 2008 and included subsections that focused on pediatric MDS and MPD.[27] This pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases was initially proposed in 2003.[10] The 2016 revision to the WHO classification has removed focus on the specific lineage (anemia, thrombocytopenia, or neutropenia) and now distinguishes cases with dysplasia in single versus multiple lineages. The category of MDS with excess blasts (MDS-EB) now encompasses the pediatric cases previously classified as refractory anemia with excess blasts (RAEB) or RAEB in transformation (RAEB-T).[28] The category of refractory cytopenia of childhood is retained as a provisional entity. The bone marrow and peripheral blood findings for MDS according to the 2008 WHO classification schema are summarized in Tables 3 and 4.[12,27] When MDS-EB is associated with the recurrent cytogenetic abnormalities that are usually associated with AML, a diagnosis of AML is made and patients are treated accordingly.
Distinguishing MDS from similar-appearing, reactive causes of dysplasia and/or cytopenias is noted to be difficult. In general, the finding of more than 10% dysplasia in a cell lineage is a diagnostic criteria for MDS; however, the 2016 WHO guidelines caution that reactive etiologies, rather than clonal, may have more than 10% dysplasia and should be excluded especially when dysplasia is subtle and/or restricted to a single lineage.[12]
The International Prognostic Scoring System is used to determine the risk of progression to AML and the outcome in adult patients with MDS. When this system was applied to children with MDS or juvenile myelomonocytic leukemia (JMML), only a blast count of less than 5% and a platelet count of more than 100 × 109 /L were associated with a better survival in MDS, and a platelet count of more than 40 × 109 /L predicted a better outcome in JMML.[29] These results suggest that MDS and JMML in children may be significantly different disorders than adult-type MDS.
Pediatric MDS can be grouped into several general categories, each with distinctive clinical and biological characteristics, as follows:[28]
- MDS arising from an inherited bone marrow failure syndrome, such as Fanconi anemia, severe congenital neutropenia, and Shwachman-Diamond syndrome.
- MDS arising from severe aplastic anemia.
- Secondary MDS arising from cytotoxic insults, such as high-dose alkylating chemotherapy.
- Primary MDS includes cases of MDS beyond those listed above, acknowledging that some of the cases characterized as primary MDS are also associated with predisposition syndromes.
Genomic characterization of pediatric primary MDS has identified specific subsets defined by alterations in selected genes. For example, germline mutations in GATA2,[30]SAMD9, or SAMD9L[31,32,33] are especially common in children with deletions of all or part of chromosome 7. Genomic characterization has also shown that primary MDS in children differs from adult MDS at the molecular level.[32,34] For more information about MDS, see the Molecular Abnormalities section.
| Type of MDS | Bone Marrow | Peripheral Blood | |
|---|---|---|---|
| a Adapted from Arber et al.[12] | |||
| b Note that cases with pancytopenia would be classified as MDS-U. | |||
| c When the marrow has <5% myeloblasts, but the peripheral blood has 2%–4% myeloblasts, the diagnosis is MDS-EB-1. | |||
| d The diagnosis of MDS-EB-2 should be made if any one of the following criteria are met: marrow with 10%–19% blasts, peripheral blood with 5%–19% blasts, or presence of Auer rods. | |||
| e Recurring chromosomal abnormalities in MDS: Unbalanced: +8, -7 or del(7q), -5 or del(5q), del(20q), -Y, i(17q) or t(17p), -13 or del(13q), del(11q), del(12p) or t(12p), del(9q), idic(X)(q13); Balanced: t(11;16)(q23;p13.3), t(3;21)(q26.2;q22.1), t(1;3)(p36.3;q21.2), t(2;11)(p21;q23), inv(3)(q21q26.2), t(6;9)(p23;q34). The WHO classification notes that the presence of these chromosomal abnormalities in presence of persistent cytopenias of undetermined origin should be considered to support a presumptive diagnosis of MDS when morphological characteristics are not observed. | |||
| f The diagnostic criteria for childhood MDS (refractory cytopenia of childhood-provisional entry) include: 1) persistent cytopenia of 1–3 cell lines with <5% bone marrow blasts, <2% peripheral blood blasts, and no ringed sideroblasts and 2) dysplastic changes in 1–3 lineages should be present. | |||
| MDS with single lineage dysplasia | Unilineage dysplasia: ≥10% in one myeloid lineage | 1–2 cytopeniasb | |
| <5% blasts | Blasts <1%c | ||
| <15% ring sideroblasts | |||
| MDS with ring sideroblasts (MDS-RS) | Erythroid dysplasia only | ||
| <5% blasts | No blasts | ||
| ≥15% ring sideroblasts | |||
| MDS with multilineage dysplasia | Dysplasia in ≥10% of cells in ≥2 myeloid lineages | 1–3 cytopenias | |
| <5% blasts | Blasts (none or <1%)c | ||
| ±15% ring sideroblasts | |||
| No Auer rods | No Auer rods | ||
| <1×109 monocytes/L | |||
| MDS with excess blasts-1 (MDS-EB-1) | Single lineage or multilineage dysplasia | Cytopenia(s) | |
| 5%–9% blastsc | <5% blastsc | ||
| No Auer rods | No Auer rods | ||
| <1×109 monocytes/L | |||
| MDS with excess blasts-2 (MDS-EB-2) | Single lineage or multilineage dysplasia | Cytopenia(s) | |
| 10%–19% blastsd | 5%–19% blastsd | ||
| Auer rods ±d | Auer rods ±d | ||
| <1×109 monocytes/L | |||
| MDS with isolated del(5q) | Normal to increased megakaryocytes (hypolobulated nuclei) | Anemia | |
| <5% blasts | Blasts (none or <1%) | ||
| No Auer rods | Normal to increased platelet count | ||
| Isolated del(5q) | |||
| MDS-unclassifiable (MDS-U) | Dysplasia in <10% of cells in ≥1 myeloid cell lineage | Cytopenias | |
| Cytogenetic abnormality associated with diagnosis of MDSe | ≤1% blastsc | ||
| <5% blasts | |||
| Provisional entity: Refractory cytopenia of childhoodf | For more information, see Table 4. | ||
| | Erythroid Lineage | Myeloid Lineage | Megakaryocyte Lineage |
|---|---|---|---|
| a Adapted from Baumann et al.[35] | |||
| b Bone marrow trephine/biopsy may be required as bone marrow in childhood refractory cytopenia of childhood is often hypocellular. | |||
| c Characteristics include abnormal nuclear lobulation, multinuclear cells, presence of nuclear bridges. | |||
| d Presence of pseudo–Pelger-Huet cells, hypo- or agranular cytoplasm, giantband forms. | |||
| e Megakaryocytes have variable size and often round or separated nuclei; the absence of megakaryocytes does not exclude the diagnosis of refractory cytopenia of childhood. | |||
| Bone Marrow Aspirateb | Dysplasia and/or megablastoid changes in ≥10% of erythroid precursorsc | Dysplasia in ≥10% of granulocytic precursors and neutrophils | Micromegakaryocytes plus other dysplastic featurese |
| <5% blastsd | |||
| Bone Marrow Biopsy | Presence of erythroid precursors | No additional criteria | Micromegakaryocytes plus other dysplastic featurese |
| Increased proerythroblasts | Immunohistochemistry positive for CD61 and CD41 | ||
| Increased number of mitoses | |||
| Peripheral Blood | Dysplasia in ≥10% of neutrophils | ||
| <2% blasts | |||
Histochemical, Immunophenotypic, and Molecular Evaluation for Childhood AML
Histochemical Evaluation
The treatment for children with acute myeloid leukemia (AML) differs significantly from that for acute lymphoblastic leukemia (ALL). As a consequence, it is critical to distinguish AML from ALL. Special histochemical stains performed on bone marrow specimens of children with acute leukemia can be helpful to confirm their diagnosis. The stains most commonly used include myeloperoxidase, periodic acid-Schiff, Sudan Black B, and esterase. In most cases, the staining pattern with these histochemical stains will distinguish AML from acute myelomonocytic leukemia (AMML) and ALL (see Table 5). Histochemical stains have been mostly replaced by flow cytometric immunophenotyping.
| | M0 | AML, APL (M1-M3) | AMML (M4) | AMoL (M5) | AEL (M6) | AMKL (M7) | ALL | |
|---|---|---|---|---|---|---|---|---|
| AEL = acute erythroid leukemia; ALL = acute lymphoblastic leukemia; AML = acute myeloid leukemia; AMKL = acute megakaryocytic leukemia; AMML = acute myelomonocytic leukemia; AMoL = acute monocytic leukemia; APL = acute promyelocytic leukemia; PAS = periodic acid-Schiff. | ||||||||
| a For more information about the morphological-histochemical classification system for AML, see the French-American-British (FAB) Classification for Childhood AMLsection. | ||||||||
| b These reactions are inhibited by fluoride. | ||||||||
| Myeloperoxidase | - | + | + | - | - | - | - | |
| Nonspecific esterases | ||||||||
| Chloracetate | - | + | + | ± | - | - | - | |
| Alpha-naphthol acetate | - | - | +b | +b | - | ±b | - | |
| Sudan Black B | - | + | + | - | - | - | - | |
| PAS | - | - | ± | ± | + | - | + | |
Immunophenotypic Evaluation
The use of monoclonal antibodies to determine cell-surface antigens of AML cells is helpful to reinforce the histologic diagnosis. Various lineage-specific monoclonal antibodies that detect antigens on AML cells should be used at the time of initial diagnostic workup, along with a battery of lineage-specific T-lymphocyte and B-lymphocyte markers to help distinguish AML from ALL and acute leukemias of ambiguous lineage. The expression of various cluster determinant (CD) proteins that are relatively lineage-specific for AML include CD33, CD13, CD14, CDw41 (or platelet antiglycoprotein IIb/IIIa), CD15, CD11B, CD36, and antiglycophorin A. Lineage-associated B-lymphocytic antigens CD10, CD19, CD20, CD22, and CD24 may be present in 10% to 20% of AML cases, but monoclonal surface immunoglobulin and cytoplasmic immunoglobulin heavy chains are usually absent; similarly, CD2, CD3, CD5, and CD7 lineage-associated T-lymphocytic antigens are present in 20% to 40% of AML cases.[36,37,38] The aberrant expression of lymphoid-associated antigens by AML cells is relatively common but generally has no prognostic significance.[36,37]
Immunophenotyping can also be helpful in distinguishing the following French-American-British (FAB) classification subtypes of AML:
- Testing for the presence of HLA-antigen D related (HLA-DR) can be helpful in identifying acute promyelocytic leukemia (APL). Overall, HLA-DR is expressed on 75% to 80% of AML cells but rarely expressed on APL cells.[39,40] In addition, APL is characterized by bright CD33 expression and by CD117 (c-KIT) expression in most cases, heterogeneous expression of CD13 with CD34, CD11a, and CD18 often negative or low.[39,40] The APL microgranular variant M3v more commonly expresses CD34 along with CD2.[39,41]
- Testing for the presence of glycoprotein Ib, glycoprotein IIb/IIIa, or Factor VIII antigen expression is helpful in making the diagnosis of M7 (megakaryocytic leukemia).
- Glycophorin expression is helpful in making the diagnosis of M6 (erythroid leukemia).
Less than 5% of cases of acute leukemia in children are of ambiguous lineage, expressing features of both myeloid and lymphoid lineage.[16,17,18] These cases are distinct from ALL with myeloid coexpression in that the predominant lineage cannot be determined by immunophenotypic and histochemical studies. The definition of leukemia of ambiguous lineage varies among studies, although most investigators now use criteria established by the European Group for the Immunological Characterization of Leukemias (EGIL) or the more stringent World Health Organization (WHO) criteria.[14,42,43] In the WHO classification, the presence of myeloperoxidase (MPO) is required to establish myeloid lineage. This is not the case for the EGIL classification. The 2016 revision to the WHO classification also denotes that in some cases, leukemia with otherwise classic B-cell ALL immunophenotype may also express low-intensity MPO without other myeloid features, and the clinical significance of that finding is unclear such that one should be cautious before designating these cases as mixed phenotype acute leukemia (MPAL).[12]
Molecular Evaluation
Molecular features of acute myeloid leukemia
Comprehensive molecular profiling of pediatric and adult AML has shown that AML is a disease demonstrating both commonalities and differences across the age spectrum.[44,45]
- Pediatric AML, in contrast to AML in adults, is typically a disease of recurring chromosomal alterations. For a list of common gene fusions, see Table 6.[44,46] Within the pediatric age range, certain gene fusions occur primarily in children younger than 5 years (e.g., NUP98, KMT2A, and CBFA2T3::GLIS2 gene fusions), while others occur primarily in children aged 5 years and older (e.g., RUNX1::RUNX1T1, CBFB::MYH11, and NPM1::RARA gene fusions).
- Pediatric patients with AML have low rates of mutations, with most cases showing less than one somatic change in protein-coding regions per megabase.[45] This mutation rate is somewhat lower than that observed in adult AML and is much lower than the mutation rate for cancers that respond to checkpoint inhibitors (e.g., melanoma).[45]
- The pattern of gene mutations differs between pediatric and adult AML cases. For example, IDH1, IDH2, TP53, RUNX1, and DNMT3A mutations are more common in adult AML than in pediatric AML, while NRAS and WT1 mutations are significantly more common in pediatric AML.[44,45]
Genetic analysis of leukemia blast cells (using both conventional cytogenetic methods and molecular methods) is performed on children with AML because both chromosomal and molecular abnormalities are important diagnostic and prognostic markers.[46,47,48,49,50] Clonal chromosomal abnormalities are identified in the blasts of about 75% of children with AML and are useful in defining subtypes with both prognostic and therapeutic significance.
Detection of molecular abnormalities can also aid in risk stratification and treatment allocation. For example, mutations of NPM and CEBPA are associated with favorable outcomes while certain mutations of FLT3 portend a high risk of relapse, and identifying the latter mutations may allow for targeted therapy.[51,52,53,54]
The 2016 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia emphasizes that recurrent chromosomal translocations in pediatric AML may be unique or have a different prevalence than in adult AML.[12] The pediatric AML chromosomal translocations that are found by conventional chromosome analysis and those that are cryptic (identified only with fluorescence in situ hybridization or molecular techniques) occur at higher rates than in adults. These recurrent translocations are summarized in Table 6.[12,45] Table 6 also shows, in the bottom three rows, additional relatively common recurrent translocations observed in children with AML.[45,48,49,55]
| Gene Fusion Product | Chromosomal Translocation | Prevalence in Pediatric AML (%) |
|---|---|---|
| a Cryptic chromosomal translocation. | ||
| KMT2A(MLL) translocated | 11q23.3 | 25.0 |
| NUP98::NSD1a | t(5;11)(q35.3;p15.5) | 7.0 |
| CBFA2T3::GLIS2a | inv(16)(p13.3;q24.3) | 3.0 |
| NUP98::KDM5Aa | t(11;12)(p15.5;p13.5) | 3.0 |
| DEK::NUP214 | t(6;9)(p22.3;q34.1) | 1.7 |
| RBM15(OTT)::MKL1(MAL) | t(1;22)(p13.3;q13.1) | 0.8 |
| MNX1::ETV6 | t(7;12)(q36.3;p13.2) | 0.8 |
| KAT6A::CREBBP | t(8;16)(p11.2;p13.3) | 0.5 |
| RUNX1::RUNX1T1 | t(8;21)(q22;q22) | 13–14 |
| CBFB::MYH11 | inv(16)(p13.1;q22) or t(16;16)(p13.1;q22) | 4–9 |
| PML::RARA | t(15;17)(q24;q21) | 6–11 |
The genomic landscape of pediatric AML cases can change from diagnosis to relapse, with mutations detectable at diagnosis dropping out at relapse and, conversely, with new mutations appearing at relapse. In a study of 20 cases for which sequencing data were available at diagnosis and relapse, a key finding was that the variant allele frequency at diagnosis strongly correlated with persistence of mutations at relapse.[56] Approximately 90% of the diagnostic variants with variant allele frequency greater than 0.4 persisted to relapse, compared with only 28% with variant allele frequency less than 0.2 (P < .001). This observation is consistent with previous results showing that presence of a mutation in the FLT3 gene resulting from internal tandem duplications (ITD) predicted for poor prognosis only when there was a high FLT3 ITD allelic ratio.
Specific recurring cytogenetic and molecular abnormalities are briefly described below. The abnormalities are listed by those in clinical use that identify patients with favorable or unfavorable prognosis, followed by other abnormalities. The nomenclature of the 2016 revision to the WHO classification of myeloid neoplasms and acute leukemia is incorporated for disease entities where relevant.
Genetic abnormalities associated with a favorable prognosis
Genetic abnormalities associated with a favorable prognosis include the following:
- Core-binding factor (CBF) AML includes cases with RUNX1::RUNX1T1 and CBFB::MYH11 gene fusions that disrupt the activity of CBF, which contains RUNX1 and CBFB. These are specific entities in the 2016 revision to the WHO classification of myeloid neoplasms and acute leukemia.
- AML with RUNX1::RUNX1T1 gene fusions (t(8;21)(q22;q22.1)). In leukemias with t(8;21), the RUNX1 (AML1) gene on chromosome 21 is fused with the RUNX1T1 (ETO) gene on chromosome 8. The t(8;21) translocation is associated with the FAB M2 subtype and with granulocytic sarcomas. Adults with t(8;21) have a more favorable prognosis than do adults with other types of AML.[47] Children with t(8;21) have a more favorable outcome than do children with AML characterized by normal or complex karyotypes,[47,57,58,59] with 5-year overall survival (OS) rates of 74% to 90%.[48,49,60] The t(8;21) translocation occurs in approximately 12% of children with AML.[48,49,60]
- AML with CBFB::MYH11 gene fusions (inv(16)(p13.1;q22) or t(16;16)(p13.1;q22)). In leukemias with inv(16), the CBFB gene at chromosome band 16q22 is fused with the MYH11 gene at chromosome band 16p13. The inv(16) translocation is associated with the FAB M4Eo subtype. Inv(16) confers a favorable prognosis for both adults and children with AML,[47,57,58,59] with a 5-year OS rate of about 85%.[48,49] Inv(16) occurs in 7% to 9% of children with AML.[48,49,60] As noted above, cases with CBFB::MYH11 fusions and cases with RUNX1::RUNX1T1 fusions have distinctive secondary mutations, with CBFB::MYH11 secondary mutations primarily restricted to genes that activate receptor tyrosine kinase signaling (NRAS, FLT3, and KIT).[61,62]
- AML with RUNX1::CBFA2T3 gene fusions (t(16;21)(q24;q22)). In leukemias with t(16;21)(q24;q22), the RUNX1 gene is fused with the CBFA2T3 gene, and the gene expression profile is closely related to that of AML cases with t(8;21) and RUNX1::RUNX1T1 fusions.[63] These patients present at a median age of 7 years and are rare, representing approximately 0.1% to 0.3% of pediatric AML cases. Among 23 patients with RUNX1::CBFA2T3 fusions, five presented with secondary AML, including two patients who had a primary diagnosis of Ewing sarcoma. Outcome for the cohort of 23 patients was favorable, with a 4-year EFS rate of 77% and a cumulative incidence of relapse rate of 0%.[63]
Both RUNX1::RUNX1T1 and CBFB::MYH11 gene fusion subtypes commonly show mutations in genes that activate receptor tyrosine kinase signaling (e.g., NRAS, FLT3, and KIT); NRAS and KIT are the most commonly mutated genes for both subtypes. The prognostic significance of activating KIT mutations in adults with CBF AML has been studied with conflicting results. A meta-analysis found that KIT mutations appear to increase the risk of relapse without an impact on OS for adults with AML and RUNX1::RUNX1T1 fusions.[64]KIT mutations are often subclonal in children and adults with CBF AML;[65,66] and in adults with AML and RUNX1::RUNX1T1 fusions, higher KIT-mutant allele ratio appears to be associated with higher risk of treatment failure.[61,65] The prognostic significance of KIT mutations in pediatric CBF AML remains unclear; some studies have found no impact of KIT mutations on outcome,[67,68,69] while other studies have reported a higher risk of treatment failure when KIT mutations are present.[66,70,71,72,73]
Although both RUNX1::RUNX1T1 and CBFB::MYH11 fusion genes disrupt the activity of CBF, cases with these genomic alterations have distinctive secondary mutations.[61,62]
- Patients with RUNX1::RUNX1T1 fusions also have frequent mutations in genes regulating chromatin conformation (e.g., ASXL1 and ASXL2) (40% of cases) and genes encoding members of the cohesin complex (20% of cases). Mutations in ASXL1 and ASXL2 and mutations in members of the cohesin complex are rare in cases with leukemia and CBFB::MYH11 fusions.[61,62]
A study of 204 adults with AML and RUNX1::RUNX1T1 fusions found that ASXL2 mutations (present in 17% of cases) and ASXL1 or ASXL2 mutations (present in 25% of cases) lacked prognostic significance.[74] Similar results, albeit with smaller numbers, were reported for children with AML and RUNX1::RUNX1T1 fusions and ASXL1 and ASXL2 mutations.[75]
- Acute promyelocytic leukemia (APL) with PML::RARA gene fusions. APL represents about 7% of children with AML.[49,76] AML with t(15;17) is invariably associated with APL, a distinct subtype of AML that is treated differently than other types of AML because of its marked sensitivity to arsenic trioxide and the differentiating effects of tretinoin. The t(15;17) translocation or other more complex chromosomal rearrangements may lead to the production of a fusion protein involving the retinoid acid receptor alpha and PML. The 2016 revision to the WHO classification does not include the t(15;17) cytogenetic designation to stress the significance of the PML::RARA fusion, which may be cryptic or result from complex karyotypic changes.[12]
Utilization of quantitative reverse transcriptase–polymerase chain reaction (RT-PCR) for PML::RARA transcripts has become standard practice.[77] Quantitative RT-PCR allows identification of the three common transcript variants and is used for monitoring response on treatment and early detection of molecular relapse. Other much less common translocations involving the retinoic acid receptor alpha can also result in APL (e.g., t(11;17)(q23;q21) involving the PLZF gene).[78,79] Identification of cases with the t(11;17)(q23;q21) is important because of their decreased sensitivity to tretinoin.[78]
- AML with mutated NPM1. NPM1 is a protein that has been linked to ribosomal protein assembly and transport as well as being a molecular chaperone involved in preventing protein aggregation in the nucleolus. Immunohistochemical methods can be used to accurately identify patients with NPM1 mutations by the demonstration of cytoplasmic localization of NPM. Mutations in the NPM1 protein that diminish its nuclear localization are primarily associated with a subset of AML with a normal karyotype, absence of CD34 expression, and an improved prognosis in the absence of FLT3 ITD mutations in adults and younger adults.[80,81,82,83,84,85]
Studies of children with AML suggest a lower rate of occurrence of NPM1 mutations in children compared with adults with normal cytogenetics. NPM1 mutations occur in approximately 8% of pediatric patients with AML and are uncommon in children younger than 2 years.[51,52,86,87]NPM1 mutations are associated with a favorable prognosis in patients with AML characterized by a normal karyotype.[51,52,87] For the pediatric population, conflicting reports have been published regarding the prognostic significance of an NPM1 mutation when a FLT3 ITD mutation is also present. One study reported that an NPM1 mutation did not completely abrogate the poor prognosis associated with having a FLT3 ITD mutation,[51,88] but other studies showed no impact of a FLT3 ITD mutation on the favorable prognosis associated with an NPM1 mutation.[45,52,87]
- AML with biallelic mutations of CEBPA. Mutations in the CEBPA gene occur in a subset of children and adults with cytogenetically normal AML.[89,90] In adults younger than 60 years, approximately 15% of cytogenetically normal AML cases have mutations in CEBPA.[84] Outcomes for adults with AML with CEBPA mutations appear to be relatively favorable and similar to that of patients with CBF leukemias.[84,91] Studies in adults with AML have demonstrated that CEBPA double-mutant, but not single-mutant, AML is independently associated with a favorable prognosis,[92,93,94,95] leading to the WHO 2016 revision that requires biallelic mutations for the disease definition.[12] However, a study of over 4,700 adults with AML found that patients with single CEBPA mutations in the bZip C-terminal domain have clinical characteristics and favorable outcomes that are similar to those of patients with double-mutant AML.[96]
CEBPA mutations occur in approximately 5% of children with AML and have been preferentially found in the cytogenetically normal subtype of AML with FAB M1 or M2.
- Patients with double CEBPA mutations or with single CEBPA bZip mutations have a median age of presentation of 12 to 13 years and have gene expression profiles that are highly related to each other.[90]
- Approximately 80% of pediatric patients have double-mutant alleles (i.e., cases with both a CEBPA TAD domain and a CEBPA bZip domain mutation), which is predictive of significantly improved survival, similar to the effect observed in adult studies.[90,97]
- In a study of nearly 3,000 children with AML, both patients with CEBPA double mutations and those with only a bZip domain mutation were observed to have a favorable prognosis, compared with patients with wild-type CEBPA.[90]
- CSF3R mutations occur in 10% to 15% of patients with CEBPA-mutated AML. When CSF3R mutations are present, they appear to be associated with an increased risk of relapse, but without an impact on overall survival.[90,98]
- In newly diagnosed patients with double-mutant CEBPA AML, germline screening should be considered in addition to usual family history queries, because 5% to 10% of these patients are reported to have a germline CEBPA mutation.[89]
- Myeloid leukemia associated with Down syndrome (GATA1 mutations). GATA1 mutations are present in most, if not all, Down syndrome children with either transient abnormal myelopoiesis (TAM) or acute megakaryoblastic leukemia (AMKL).[99,100,101,102]GATA1 mutations were also observed in 9% of non–Down syndrome children and 4% of adults with AMKL (with coexistence of amplification of the RCAN1 [DSCR1] gene on chromosome 21 in 9 of 10 cases).[103]GATA1 is a transcription factor that is required for normal development of erythroid cells, megakaryocytes, eosinophils, and mast cells.
GATA1 mutations confer increased sensitivity to cytarabine by down-regulating cytidine deaminase expression, possibly providing an explanation for the superior outcome of children with Down syndrome and M7 AML when treated with cytarabine-containing regimens.[104]
Genetic abnormalities associated with an unfavorable prognosis
Genetic abnormalities associated with an unfavorable prognosis include the following:
- Chromosomes 5 and 7. Chromosomal abnormalities associated with poor prognosis in adults with AML include those involving chromosome 5 (del(5q)) and chromosome 7 (monosomy 7).[47,105,106] These cytogenetic subgroups represent approximately 2% and 4% of pediatric AML cases, respectively, and are also associated with poor prognosis in children.[48,105,106,107,108,109]
In the past, patients with del(7q) were also considered to be at high risk of treatment failure, and data from adults with AML support a poor prognosis for both del(7q) and monosomy 7.[50] However, outcome for children with del(7q), but not monosomy 7, appears comparable to that of other children with AML.[49,108] The presence of del(7q) does not abrogate the prognostic significance of favorable cytogenetic characteristics (e.g., inv(16) and t(8;21)).[47,108]
Chromosome 5 and 7 abnormalities appear to lack prognostic significance in AML patients with Down syndrome who are aged 4 years and younger.[110]
- Hypodiploidy. Hypodiploidy is defined as a modal chromosome number of less than or equal to 45. This occurs rarely in pediatric patients with AML. In a retrospective cohort analysis, the International Berlin-Frankfurt-Münster AML Study Group aimed to characterize hypodiploidy in pediatric patients with AML. The study excluded several patient groups, including patients with APL, Down syndrome, or loss of chromosome 7.[111] Their observations included the following:
- Hypodiploidy was observed in 1.3% of children with AML. Approximately 80% of patients had a modal chromosome number of 45, and the remaining 20% of patients had a modal chromosome number of either 43 or 44.
- Most patients (>80%) with a modal chromosome number of 43 or 44 also met the criteria for complex karyotype. In this study, a complex karyotype was defined as at least three independent chromosomal abnormalities, regardless of whether these were structural abnormalities or defects in chromosome number, and an absence of recurrent aberrations as defined by the WHO.
- Patients with a modal chromosome number of 43 or 44 had decreased EFS rates and OS rates when compared with patients who had 45 chromosomes (EFS rate, 21% vs. 37%; P = .07; OS rate, 33% vs. 56%; P = .1).
- AML with GATA2 or MECOM (inv(3)(q21.3;q26.2) or t(3;3)(q21.3;q26.2)). MECOM at chromosome 3q26 codes for two proteins, EVI1 and MDS1::EVI1, both of which are transcription regulators. The inv(3) and t(3;3) abnormalities lead to overexpression of EVI1 and to reduced expression of GATA2.[112,113] These abnormalities are associated with poor prognosis in adults with AML,[47,105,114] but are very uncommon in children (<1% of pediatric AML cases).[48,58,115]
Abnormalities involving MECOM can be detected in some AML cases with other 3q abnormalities and are also associated with poor prognosis.
- FLT3 mutations. Presence of a FLT3 ITD mutation appears to be associated with poor prognosis in adults with AML,[116] particularly when both alleles are mutated or there is a high ratio of the mutant allele to the normal allele.[117]FLT3 ITD mutations also convey a poor prognosis in children with AML.[54,88,118,119,120] The frequency of FLT3 ITD mutations in children is lower than that observed in adults, especially for children younger than 10 years, for whom 5% to 10% of cases have the mutation (compared with approximately 30% in adults).[119,120]
The prognostic significance of FLT3 ITD is modified by the presence of other recurring genomic alterations. The prevalence of FLT3 ITD is increased in certain genomic subtypes of pediatric AML, including those with the NUP98::NSD1 fusion gene, of which 80% to 90% have FLT3 ITD.[121,122] Approximately 15% of patients with FLT3 ITD have NUP98::NSD1 fusions, and patients with both FLT3 ITD and NUP98::NSD1 fusions have a poorer prognosis than do patients who have FLT3 ITD without NUP98::NSD1 fusions.[122] For patients who have FLT3 ITD, the presence of either WT1 mutations or NUP98::NSD1 fusions is associated with poorer outcome (EFS rates below 25%) than for patients who have FLT3 ITD without these alterations.[45] Conversely, when FLT3 ITD is accompanied by NPM1 mutations, the outcome is relatively favorable and is similar to that of pediatric AML cases without FLT3 ITD.[45]
For APL, FLT3 ITD and point mutations occur in 30% to 40% of children and adults.[117,119,123,124] Presence of the FLT3 ITD mutation is strongly associated with the microgranular variant (M3v) of APL and with hyperleukocytosis.[123,125,126,127] It remains unclear whether FLT3 mutations are associated with poorer prognosis in patients with APL who are treated with modern therapy that includes tretinoin and arsenic trioxide.[124,126,128,129,130,131]
Activating point mutations of FLT3 have also been identified in both adults and children with AML, although the clinical significance of these mutations is not clearly defined. Some of these point mutations appear to be specific to pediatric patients.[45]
- AML with t(16;21)(p11;q22); FUS::ERG gene fusions. In leukemias with t(16;21)(p11;q22), the FUS gene is joined with the ERG gene, producing a distinctive AML subtype with a gene expression profile that clusters separately from other cytogenetic subgroups.[63] These patients present at a median age of 8 to 9 years and are rare, representing approximately 0.3% to 0.5% of pediatric AML cases. For a cohort of 31 patients with AML and FUS::ERG fusions, outcome was poor, with a 4-year EFS rate of 7% and a cumulative incidence of relapse rate of 74%.[63]
Other genetic abnormalities observed in pediatric AML
Other genetic abnormalities observed in pediatric AML include the following:
- KMT2A (MLL) gene rearrangements. KMT2A gene rearrangement occurs in approximately 20% of children with AML.[48,49] These cases, including most AMLs secondary to epipodophyllotoxin exposure,[132] are generally associated with monocytic differentiation (FAB M4 and M5). KMT2A rearrangements are also reported in approximately 10% of FAB M7 (AMKL) patients (see below).[103,133]
The most common translocation, representing approximately 50% of KMT2A-rearranged cases in the pediatric AML population, is t(9;11)(p22;q23), in which the KMT2A gene is fused with the MLLT3 gene.[134] The 2016 revision to the WHO classification defined AML with t(9;11)(p21.3;q23.3); MLLT3::KMT2A gene fusions as a distinctive disease entity. However, more than 50 different fusion partners have been identified for the KMT2A gene in patients with AML.
The median age for 11q23/KMT2A-rearranged cases in children is approximately 2 years, and most translocation subgroups have a median age at presentation of younger than 5 years.[134] However, significantly older median ages are seen at presentation of pediatric cases with t(6;11)(q27;q23) (12 years) and t(11;17)(q23;q21) (9 years).[134]
Outcome for patients with de novo AML and KMT2A gene rearrangements is generally reported as being similar to or slightly worse than the outcome observed in other patients with AML.[47,48,134,135,136] As the KMT2A gene can participate in translocations with many different fusion partners, the specific fusion partner appears to influence prognosis, as demonstrated by a large international retrospective study evaluating outcome for 756 children with 11q23- or KMT2A-rearranged AML.[134,136] This was also seen in patients in the COG AAML0531 (NCT00372593) trial (n = 215), which resulted in a wide range of outcomes.[136] This overall less-favorable outcome was abrogated in one arm of the AAML0531 trial, in patients whose treatment included gemtuzumab ozogamicin. The EFS rate for patients with KMT2A-rearranged AML was superior with gemtuzumab ozogamicin treatment (EFS rate, 48% with gemtuzumab ozogamicin vs. 29% without; P = .003). Outcomes for patients with KMT2A-rearranged AML who received gemtuzumab ozogamicin are similar to the outcomes observed in patients without KMT2A-rearrangements.[136]
For patients with the most prevalent KMT2A-rearranged subtype of AML, t(9;11)(p21.3;q23.3)/MLLT3::KMT2A fusions, single clinical trial groups have variably described a more favorable prognosis; however, neither the international retrospective study nor the COG study confirmed the favorable prognosis for this subgroup.[47,48,134,136] Furthermore, an international collaboration evaluating pediatric AMKL patients observed that the presence of t(9;11), which was seen in approximately 5% of AMKL cases, was associated with an inferior outcome compared with other AMKL cases.[133]
KMT2A-rearranged AML subgroups that are associated with poor outcome include the following:
- Cases with the t(10;11) translocation are a group at high risk of relapse in bone marrow and the CNS.[47,49] Some cases with the t(10;11) translocation have fusion of the KMT2A gene with the MLLT10 at 10p12, while others have fusion of KMT2A with ABI1 at 10p11.2. An international retrospective study found that these cases, which present at a median age of approximately 1 to 3 years, have a 5-year EFS rate of 17% to 30%.[134,136]
- Patients with t(6;11)(q27;q23) have a poor outcome, with a 5-year EFS rate of 11% to 15%.[136]
- Patients with t(4;11)(q21;q23) often present with hyperleukocytosis and also have a poor outcome, with a 5-year EFS rate of 0% to 29%.[134,136]
- Patients with t(11;19)(q23;p13.3) have a poor outcome, with a 5-year EFS rate of 14%.[136]
- A follow-up study by the international collaborative group demonstrated that additional cytogenetic abnormalities further influenced outcome of children with KMT2A translocations, with complex karyotypes and trisomy 19 predicting poor outcome and trisomy 8 predicting a more favorable outcome.[137]
- The addition of gemtuzumab ozogamicin therapy improved the poor outcome of these patients with KMT2A-rearranged high-risk translocation partners (27% [95% confidence interval (CI), 14%–41%] vs. 6% [95% CI, 1%–18%]; P = .013).[136]
- AML with DEK::NUP214 (t(6;9)(p23;q34.1)) gene fusions. t(6;9) leads to the formation of a leukemia-associated fusion protein DEK::NUP214.[138,139] This subgroup of AML has been associated with a poor prognosis in adults with AML,[138,140,141] and occurs infrequently in children (less than 1% of AML cases). The median age of children with AML and DEK::NUP214 fusions is 10 to 11 years, and approximately 40% of pediatric patients have FLT3 ITD.[142,143]
t(6;9) AML appears to be associated with a high risk of treatment failure in children, particularly for those not proceeding to allogeneic stem cell transplantation.[48,139,142,143]
- Molecular subgroups of non–Down syndrome acute megakaryoblastic leukemia (AMKL). AMKL accounts for approximately 10% of pediatric AML and includes substantial heterogeneity at the molecular level. Molecular subtypes of AMKL are listed below.
- CBFA2T3::GLIS2 gene fusions. CBFA2T3::GLIS2 is a fusion resulting from a cryptic chromosome 16 inversion (inv(16)(p13.3;q24.3)).[144,145,146,147,148] It occurs commonly in non–Down syndrome AMKL, representing 16% to 27% of pediatric AMKL and presenting with a median age of 1 year.[103,146,149,150] It appears to be associated with unfavorable outcome.[103,144,148,149,150]
In a study of approximately 2,000 children with AML, the CBFA2T3::GLIS2 fusion was identified in 39 cases (1.9%), with a median age at presentation of 1.5 years, and with all cases observed in children younger than 3 years.[151] Approximately one-half of cases had M7 megakaryoblastic morphology, and 29% of patients were Black or African American (exceeding the 12.8% frequency in patients lacking the fusion). The CBFA2T3::GLIS2 fusion was an independent prognostic factor for both OS and EFS. The OS rate at 5 years was 22% for patients with CBFA2T3::GLIS2 fusions versus 63% for fusion-negative patients. Leukemia cells with CBFA2T3::GLIS2 fusions have a distinctive immunophenotype (initially reported as the RAM phenotype),[152,153] with high CD56, dim or negative expression of CD45 and CD38, and a lack of HLA-DR expression.
- KMT2A rearrangements. Cases with KMT2A translocations represent 10% to 17% of pediatric AMKL, with MLLT3 being the most common KMT2A fusion partner.[103,133,149] Patients with KMT2A rearrangements appear to have an inferior outcome among children with AMKL, with OS rates at 4 to 5 years of approximately 30%.[103,133,149] An international collaboration evaluating pediatric AMKL observed that the presence of t(9;11)/MLLT3::KMT2A fusions, which was seen in approximately 5% of AMKL cases (n = 21), was associated with an inferior outcome (5-year OS rate, approximately 20%) compared with other AMKL cases and other KMT2A rearrangements (n = 17), each with a 5-year OS rate of 50% to 55%.[133] Inferior outcome was not observed for patients (n = 17) with other KMT2A rearrangements.
- NUP98::KDM5A gene fusions. NUP98::KDM5A fusions are observed in approximately 10% of pediatric AMKL cases [103,149] and is seen at lower rates in non-AMKL cases.[150] However, approximately two-thirds of children with NUP98::KDM5A fusions have a non-AMKL FAB subtype (see below).[154] Patients with NUP98::KDM5A fusions showed a trend towards inferior prognosis, although the small number of cases studied limits confidence in this assessment.[103,149]
- RBM15::MKL1 gene fusions. The t(1;22)(p13;q13) translocation that produces RBM15::MKL1 fusions is uncommon (<1% of pediatric AML) and is restricted to acute megakaryocytic leukemia (AMKL).[48,150,155,156,157,158] Studies have found that t(1;22)(p13;q13) is observed in 10% to 18% of children with AMKL who have evaluable cytogenetics or molecular genetics.[103,133,149] Most AMKL cases with t(1;22) occur in infants, with the median age at presentation (4–7 months) being younger than that for other children with AMKL.[133,146,159] Cases with detectable RBM15::MKL1 fusion transcripts in the absence of t(1;22) have also been reported because these young patients usually have hypoplastic bone marrow.[156]
An international collaborative retrospective study of 51 t(1;22) cases reported that patients with this abnormality had a 5-year EFS rate of 54.5% and an OS rate of 58.2%, similar to the rates for other children with AMKL.[133] In another international retrospective analysis of 153 cases with non–Down syndrome AMKL who had samples available for molecular analysis, the 4-year EFS rate for patients with t(1;22) was 59% and the OS rate was 70%, significantly better than AMKL patients with other specific genetic abnormalities (CBFA2T3::GUS2 fusions, NUP98::KDM5A fusions, KMT2A rearrangements, monosomy 7).[149]
- HOX rearrangements. Cases with a gene fusion involving a HOX cluster gene represented 15% of pediatric AMKL in one report.[103] This report observed that these patients appear to have a relatively favorable prognosis, although the small number of cases studied limits confidence in this assessment.
- GATA1 mutations. GATA1-truncating mutations in non–Down syndrome AMKL arise in young children (median age, 1–2 years) and are associated with amplification of the RCAN1 (DSCR1) gene on chromosome 21.[103] These patients represented approximately 10% of non–Down syndrome AMKL and appeared to have a favorable outcome if there were no prognostically unfavorable fusion genes also present, although the number of patients studied was small (n = 8).[103]
- CBFA2T3::GLIS2 gene fusions. CBFA2T3::GLIS2 is a fusion resulting from a cryptic chromosome 16 inversion (inv(16)(p13.3;q24.3)).[144,145,146,147,148] It occurs commonly in non–Down syndrome AMKL, representing 16% to 27% of pediatric AMKL and presenting with a median age of 1 year.[103,146,149,150] It appears to be associated with unfavorable outcome.[103,144,148,149,150]
- MYST3::CREBBP (t(8;16)). The t(8;16) translocation fuses the MYST3 gene on chromosome 8p11 to CREBBP on chromosome 16p13. t(8;16) AML rarely occurs in children. In an International Berlin-Frankfurt-Münster (iBFM) AML study of 62 children, presence of this translocation was associated with younger age at diagnosis (median, 1.2 years), FAB M4/M5 phenotype, erythrophagocytosis, leukemia cutis, and disseminated intravascular coagulation.[160] Outcome for children with t(8;16) AML appears similar to other types of AML.
A substantial proportion of infants diagnosed with t(8;16) AML in the first month of life show spontaneous remission, although AML recurrence may occur months to years later.[160,161,162,163] These observations suggest that a watch and wait policy could be considered in cases of t(8;16) AML diagnosed in the neonatal period if close long-term monitoring can be ensured.[160]
- t(7;12)(q36;p13). The t(7;12)(q36;p13) translocation involves ETV6 on chromosome 12p13 and variable breakpoints on chromosome 7q36 in the region of MNX1. The translocation may be cryptic by conventional karyotyping and in some cases may be confirmed only by FISH.[164,165] This alteration occurs virtually exclusively in children younger than 2 years, is mutually exclusive with the KMT2A rearrangement, and is associated with a high risk of treatment failure.[48,49,87,164,166,167]
- NUP98 gene fusions. NUP98 has been reported to form leukemogenic gene fusions with more than 20 different partners.[168] In the pediatric AML setting, the two most common gene fusions are NUP98::NSD1 and NUP98::KDM5A, with the former observed in one report in approximately 15% of cytogenetically normal pediatric AML and the latter observed in approximately 10% of pediatric AMKL (see above).[103,121,146] AML cases with either NUP98 gene fusion show high expression of HOXA and HOXB genes, indicative of a stem cell phenotype.[139,146]
The NUP98::NSD1 gene fusion, which is often cytogenetically cryptic, results from the fusion of NUP98 (chromosome 11p15) with NSD1 (chromosome 5q35).[121,122,139,169] This alteration occurs in approximately 4% to 7% of pediatric AML cases.[12,55,121,139,170]
- The highest frequency of NUP98::NSD1 fusions in the pediatric population is observed in children aged 5 to 9 years (approximately 8%), with a lower frequency in younger children (approximately 2% in children younger than 2 years).
- Patients with NUP98::NSD1 fusions present with a high white blood cell (WBC) count (median, 147 × 109 /L in one study).[121,122] Most patients with AML and NUP98::NSD1 fusions do not show cytogenetic aberrations.[121,139]
- A high percentage of patients with NUP98::NSD1 fusions (74%–90%) have FLT3 ITD.[55,121,122]
- A study that included 12 children with AML and NUP98::NSD1 fusions reported that although all patients achieved a complete response (CR), the presence of NUP98::NSD1 fusions independently predicted poor prognosis. Children with AML and NUP98::NSD1 fusions had a high risk of relapse, with a resulting 4-year EFS rate of approximately 10%.[121] In another study that included children (n = 38) and adults (n = 7) with AML and NUP98::NSD1 fusions, presence of both NUP98::NSD1 fusions and FLT3 ITD independently predicted poor prognosis; patients with both lesions had a low CR rate (approximately 30%) and a low 3-year EFS rate (approximately 15%).[122]
- In a study of children with refractory AML, NUP98 was overrepresented compared with a cohort who did achieve remission (21% [6 of 28 patients] vs. <4%).[171]
The NUP98::KDM5A gene fusion results from the fusion of the NUP98 gene with the KDM5A gene, which results from a cytogenetically cryptic translocation, t(11;12)(p15;p13).[172] Approximately 2% of pediatric AML patients have NUP98::KDM5A fusions, and these cases tend to present at a young age (median age, 3 years).[154]
- Cases with NUP98::KDM5A fusions tend to be AMKL (34%), followed by FAB M5 (21%), and FAB M6 (17%).[154]NUP98::KDM5A fusions are observed in approximately 10% of pediatric AMKL cases.[103,149]
- Other genetic aberrations associated with pediatric AML, including FLT3 mutations, are uncommon in patients with NUP98::KDM5A fusions.[154]
- Prognosis for children with NUP98::KDM5A fusions is inferior to that of other children with AML (5-year EFS rate of 29.6% ± 14.6% and an OS rate of 34.1% ± 16.1%).[154]
- RUNX1 mutations. AML with mutated RUNX1, which is a provisional entity in the 2016 WHO classification of AML and related neoplasms, is more common in adults than in children. In adults, the RUNX1 mutation is associated with a high risk of treatment failure. In a study of children with AML, RUNX1 mutations were observed in 11 of 503 patients (approximately 2%). Six of 11 patients with RUNX1-mutated AML failed to achieve remission and their 5-year EFS rate was 9%, suggesting that the RUNX1 mutation confers a poor prognosis in both children and adults.[173]
- RAS mutations. Although mutations in RAS have been identified in 20% to 25% of patients with AML, the prognostic significance of these mutations has not been clearly shown.[87,174] Mutations in NRAS are observed more commonly than mutations in KRAS in pediatric AML cases.[87,175]RAS mutations occur with similar frequency for all Type II alteration subtypes, with the exception of APL, for which RAS mutations are seldom observed.[87]
- KIT mutations. Mutations in KIT occur in approximately 5% of AML, but in 10% to 40% of AML with CBF abnormalities.[73,87,175,176]
The prognostic significance of activating KIT mutations in adults with CBF AML has been studied with conflicting results. A meta-analysis found that KIT mutations appear to increase the risk of relapse without an impact on OS for adults with AML and RUNX1::RUNX1T1 fusions.[64]KIT mutations are often subclonal in children and adults with CBF AML;[65,66] in adults with AML and RUNX1::RUNX1T1 mutations, higher KIT mutant–allele ratio appears to be associated with higher risk of treatment failure.[61,65] The prognostic significance of KIT mutations in pediatric CBF AML remains unclear; some studies found no impact of KIT mutations on outcome,[67,68,69] while other studies reported a higher risk of treatment failure when KIT mutations were present.[66,70,71,72,73]
- WT1 mutations. WT1, a zinc-finger protein regulating gene transcription, is mutated in approximately 10% of cytogenetically normal cases of AML in adults.[177,178,179,180] The WT1 mutation has been shown in some,[177,178,180] but not all studies [179] to be an independent predictor of worse disease-free survival, EFS, and OS of adults.
In children with AML, WT1 mutations are observed in approximately 10% of cases.[181,182] Cases with WT1 mutations are enriched among children with normal cytogenetics and FLT3 ITD, but are less common among children younger than 3 years.[181,182] AML cases with NUP98::NSD1 fusions are enriched for both FLT3 ITD and WT1 mutations.[121] In univariate analyses, WT1 mutations are predictive of poorer outcome in pediatric patients, but the independent prognostic significance of WT1 mutation status is unclear because of its strong association with FLT3 ITD and its association with NUP98::NSD1 fusions.[121,181,182] The largest study of WT1 mutations in children with AML observed that children with WT1 mutations in the absence of FLT3 ITD had outcomes similar to that of children without WT1 mutations, while children with both WT1 mutation and FLT3 ITD had survival rates less than 20%.[181]
In a study of children with refractory AML, WT1 was overrepresented compared with a cohort who did achieve remission (54% [15 of 28 patients] vs. 15%).[171]
- DNMT3A mutations. Mutations of the DNMT3A gene have been identified in approximately 20% of adult AML patients and are uncommon in patients with favorable cytogenetics but occur in one-third of adult patients with intermediate-risk cytogenetics.[183] Mutations in this gene are independently associated with poor outcome.[183,184,185]DNMT3A mutations are virtually absent in children.[186]
- IDH1 and IDH2 mutations. Mutations in IDH1 and IDH2, which code for isocitrate dehydrogenase, occur in approximately 20% of adults with AML,[187,188,189,190,191] and they are enriched in patients with NPM1 mutations.[188,189,192] The specific mutations that occur in IDH1 and IDH2 create a novel enzymatic activity that promotes conversion of alpha-ketoglutarate to 2-hydroxyglutarate.[193,194] This novel activity appears to induce a DNA hypermethylation phenotype similar to that observed in AML cases with loss of function mutations in TET2.[192]
Mutations in IDH1 and IDH2 are rare in pediatric AML, occurring in 0% to 4% of cases.[186,195,196,197,198,199] There is no indication of a negative prognostic effect for IDH1 and IDH2 mutations in children with AML.[195]
- CSF3R mutations. CSF3R is the gene encoding the granulocyte colony-stimulating factor (G-CSF) receptor, and activating mutations in CSF3R are observed in 2% to 3% of pediatric AML cases.[200] These mutations lead to enhanced signaling through the G-CSF receptor, and they are primarily observed in AML with either CEBPA mutations or with CBF abnormalities (RUNX1::RUNX1T1 and CBFB::MYH11 fusions).[200] In a study of 2,150 pediatric patients with AML, 35 patients (1.6%) were found to have CSF3R mutations; 30 (89%) of these cases were in patients with either RUNX1::RUNX1T1 fusions (n = 18) or with CEBPA mutations (n = 12).[98] Risk of relapse was significantly higher for patients with co-occurring CSF3R and CEBPA mutations compared with patients with RUNX1::RUNX1T1 fusions and CSF3R mutations.[98] Although relapse rates are higher in patients with AML that have co-occurring CSF3R and CEBPA mutations, overall survival is not adversely impacted, reflecting a high salvage rate with reinduction therapy and stem cell transplant.[90]
Activating mutations in CSF3R are also observed in patients with severe congenital neutropenia. These mutations are not the cause of severe congenital neutropenia, but rather arise as somatic mutations and can represent an early step in the pathway to AML.[201] In one study of patients with severe congenital neutropenia, 34% of patients who had not developed a myeloid malignancy had CSF3R mutations detectable in peripheral blood neutrophils and mononuclear cells, while 78% of patients who had developed a myeloid malignancy showed CSF3R mutations.[201] A study of 31 patients with severe congenital neutropenia who developed AML or MDS observed CSF3R mutations in approximately 80% of patients, and also observed a high frequency of RUNX1 mutations (approximately 60%), suggesting cooperation between CSF3R and RUNX1 mutations for leukemia development within the context of severe congenital neutropenia.[202]
References:
- Bennett JM, Catovsky D, Daniel MT, et al.: Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 33 (4): 451-8, 1976.
- Bennett JM, Catovsky D, Daniel MT, et al.: Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med 103 (4): 620-5, 1985.
- Bennett JM, Catovsky D, Daniel MT, et al.: Criteria for the diagnosis of acute leukemia of megakaryocyte lineage (M7). A report of the French-American-British Cooperative Group. Ann Intern Med 103 (3): 460-2, 1985.
- Bennett JM, Catovsky D, Daniel MT, et al.: A variant form of hypergranular promyelocytic leukaemia (M3) Br J Haematol 44 (1): 169-70, 1980.
- Cheson BD, Bennett JM, Kopecky KJ, et al.: Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 21 (24): 4642-9, 2003.
- Bennett JM, Catovsky D, Daniel MT, et al.: Proposal for the recognition of minimally differentiated acute myeloid leukaemia (AML-MO) Br J Haematol 78 (3): 325-9, 1991.
- Kaleem Z, White G: Diagnostic criteria for minimally differentiated acute myeloid leukemia (AML-M0). Evaluation and a proposal. Am J Clin Pathol 115 (6): 876-84, 2001.
- Vardiman JW, Harris NL, Brunning RD: The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 100 (7): 2292-302, 2002.
- Jaffe ES, Harris NL, Stein H, et al., eds.: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press, 2001. World Health Organization Classification of Tumours, 3.
- Hasle H, Niemeyer CM, Chessells JM, et al.: A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia 17 (2): 277-82, 2003.
- Arber DA, Vardiman JW, Brunning RD: Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 110-23.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.
- Borowitz MJ, Béné MC, Harris NL: Acute leukaemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 150-5.
- Alexander TB, Gu Z, Iacobucci I, et al.: The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 562 (7727): 373-379, 2018.
- Gerr H, Zimmermann M, Schrappe M, et al.: Acute leukaemias of ambiguous lineage in children: characterization, prognosis and therapy recommendations. Br J Haematol 149 (1): 84-92, 2010.
- Rubnitz JE, Onciu M, Pounds S, et al.: Acute mixed lineage leukemia in children: the experience of St Jude Children's Research Hospital. Blood 113 (21): 5083-9, 2009.
- Al-Seraihy AS, Owaidah TM, Ayas M, et al.: Clinical characteristics and outcome of children with biphenotypic acute leukemia. Haematologica 94 (12): 1682-90, 2009.
- Matutes E, Pickl WF, Van't Veer M, et al.: Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood 117 (11): 3163-71, 2011.
- Hrusak O, de Haas V, Stancikova J, et al.: International cooperative study identifies treatment strategy in childhood ambiguous lineage leukemia. Blood 132 (3): 264-276, 2018.
- Orgel E, Alexander TB, Wood BL, et al.: Mixed-phenotype acute leukemia: A cohort and consensus research strategy from the Children's Oncology Group Acute Leukemia of Ambiguous Lineage Task Force. Cancer 126 (3): 593-601, 2020.
- Bennett JM, Catovsky D, Daniel MT, et al.: Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 51 (2): 189-99, 1982.
- Mandel K, Dror Y, Poon A, et al.: A practical, comprehensive classification for pediatric myelodysplastic syndromes: the CCC system. J Pediatr Hematol Oncol 24 (7): 596-605, 2002.
- Bennett JM: World Health Organization classification of the acute leukemias and myelodysplastic syndrome. Int J Hematol 72 (2): 131-3, 2000.
- Head DR: Proposed changes in the definitions of acute myeloid leukemia and myelodysplastic syndrome: are they helpful? Curr Opin Oncol 14 (1): 19-23, 2002.
- Nösslinger T, Reisner R, Koller E, et al.: Myelodysplastic syndromes, from French-American-British to World Health Organization: comparison of classifications on 431 unselected patients from a single institution. Blood 98 (10): 2935-41, 2001.
- Brunning RD, Porwit A, Orazi A, et al.: Myelodysplastic syndromes/neoplasms overview. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 88-93.
- Wlodarski MW, Sahoo SS, Niemeyer CM: Monosomy 7 in Pediatric Myelodysplastic Syndromes. Hematol Oncol Clin North Am 32 (4): 729-743, 2018.
- Hasle H, Baumann I, Bergsträsser E, et al.: The International Prognostic Scoring System (IPSS) for childhood myelodysplastic syndrome (MDS) and juvenile myelomonocytic leukemia (JMML). Leukemia 18 (12): 2008-14, 2004.
- Wlodarski MW, Hirabayashi S, Pastor V, et al.: Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 127 (11): 1387-97; quiz 1518, 2016.
- Narumi S, Amano N, Ishii T, et al.: SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet 48 (7): 792-7, 2016.
- Schwartz JR, Ma J, Lamprecht T, et al.: The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun 8 (1): 1557, 2017.
- Davidsson J, Puschmann A, Tedgård U, et al.: SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 32 (5): 1106-1115, 2018.
- Pastor V, Hirabayashi S, Karow A, et al.: Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia 31 (3): 759-762, 2017.
- Baumann I, Niemeyer CM, Bennett JM, et al.: Childhood myelodysplastic syndrome. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 104-7.
- Kuerbitz SJ, Civin CI, Krischer JP, et al.: Expression of myeloid-associated and lymphoid-associated cell-surface antigens in acute myeloid leukemia of childhood: a Pediatric Oncology Group study. J Clin Oncol 10 (9): 1419-29, 1992.
- Smith FO, Lampkin BC, Versteeg C, et al.: Expression of lymphoid-associated cell surface antigens by childhood acute myeloid leukemia cells lacks prognostic significance. Blood 79 (9): 2415-22, 1992.
- Dinndorf PA, Andrews RG, Benjamin D, et al.: Expression of normal myeloid-associated antigens by acute leukemia cells. Blood 67 (4): 1048-53, 1986.
- Zhou Y, Jorgensen JL, Wang SA, et al.: Usefulness of CD11a and CD18 in flow cytometric immunophenotypic analysis for diagnosis of acute promyelocytic leukemia. Am J Clin Pathol 138 (5): 744-50, 2012.
- Paietta E, Goloubeva O, Neuberg D, et al.: A surrogate marker profile for PML/RAR alpha expressing acute promyelocytic leukemia and the association of immunophenotypic markers with morphologic and molecular subtypes. Cytometry B Clin Cytom 59B (1): 1-9, 2004.
- Lin P, Hao S, Medeiros LJ, et al.: Expression of CD2 in acute promyelocytic leukemia correlates with short form of PML-RARalpha transcripts and poorer prognosis. Am J Clin Pathol 121 (3): 402-7, 2004.
- Bene MC, Castoldi G, Knapp W, et al.: Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 9 (10): 1783-6, 1995.
- Vardiman JW, Thiele J, Arber DA, et al.: The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114 (5): 937-51, 2009.
- Tarlock K, Meshinchi S: Pediatric acute myeloid leukemia: biology and therapeutic implications of genomic variants. Pediatr Clin North Am 62 (1): 75-93, 2015.
- Bolouri H, Farrar JE, Triche T, et al.: The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 24 (1): 103-112, 2018.
- Creutzig U, van den Heuvel-Eibrink MM, Gibson B, et al.: Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 120 (16): 3187-205, 2012.
- Grimwade D, Walker H, Oliver F, et al.: The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood 92 (7): 2322-33, 1998.
- Harrison CJ, Hills RK, Moorman AV, et al.: Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J Clin Oncol 28 (16): 2674-81, 2010.
- von Neuhoff C, Reinhardt D, Sander A, et al.: Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J Clin Oncol 28 (16): 2682-9, 2010.
- Grimwade D, Hills RK, Moorman AV, et al.: Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 116 (3): 354-65, 2010.
- Brown P, McIntyre E, Rau R, et al.: The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood 110 (3): 979-85, 2007.
- Hollink IH, Zwaan CM, Zimmermann M, et al.: Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia 23 (2): 262-70, 2009.
- Ho PA, Alonzo TA, Gerbing RB, et al.: Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): a report from the Children's Oncology Group. Blood 113 (26): 6558-66, 2009.
- Meshinchi S, Alonzo TA, Stirewalt DL, et al.: Clinical implications of FLT3 mutations in pediatric AML. Blood 108 (12): 3654-61, 2006.
- Struski S, Lagarde S, Bories P, et al.: NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia 31 (3): 565-572, 2017.
- Farrar JE, Schuback HL, Ries RE, et al.: Genomic Profiling of Pediatric Acute Myeloid Leukemia Reveals a Changing Mutational Landscape from Disease Diagnosis to Relapse. Cancer Res 76 (8): 2197-205, 2016.
- Creutzig U, Zimmermann M, Ritter J, et al.: Definition of a standard-risk group in children with AML. Br J Haematol 104 (3): 630-9, 1999.
- Raimondi SC, Chang MN, Ravindranath Y, et al.: Chromosomal abnormalities in 478 children with acute myeloid leukemia: clinical characteristics and treatment outcome in a cooperative pediatric oncology group study-POG 8821. Blood 94 (11): 3707-16, 1999.
- Lie SO, Abrahamsson J, Clausen N, et al.: Treatment stratification based on initial in vivo response in acute myeloid leukaemia in children without Down's syndrome: results of NOPHO-AML trials. Br J Haematol 122 (2): 217-25, 2003.
- Klein K, Kaspers G, Harrison CJ, et al.: Clinical Impact of Additional Cytogenetic Aberrations, cKIT and RAS Mutations, and Treatment Elements in Pediatric t(8;21)-AML: Results From an International Retrospective Study by the International Berlin-Frankfurt-Münster Study Group. J Clin Oncol 33 (36): 4247-58, 2015.
- Duployez N, Marceau-Renaut A, Boissel N, et al.: Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood 127 (20): 2451-9, 2016.
- Faber ZJ, Chen X, Gedman AL, et al.: The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet 48 (12): 1551-1556, 2016.
- Noort S, Zimmermann M, Reinhardt D, et al.: Prognostic impact of t(16;21)(p11;q22) and t(16;21)(q24;q22) in pediatric AML: a retrospective study by the I-BFM Study Group. Blood 132 (15): 1584-1592, 2018.
- Chen W, Xie H, Wang H, et al.: Prognostic Significance of KIT Mutations in Core-Binding Factor Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis. PLoS One 11 (1): e0146614, 2016.
- Christen F, Hoyer K, Yoshida K, et al.: Genomic landscape and clonal evolution of acute myeloid leukemia with t(8;21): an international study on 331 patients. Blood 133 (10): 1140-1151, 2019.
- Tarlock K, Alonzo TA, Wang YC, et al.: Functional Properties of KIT Mutations Are Associated with Differential Clinical Outcomes and Response to Targeted Therapeutics in CBF Acute Myeloid Leukemia. Clin Cancer Res 25 (16): 5038-5048, 2019.
- Shih LY, Liang DC, Huang CF, et al.: Cooperating mutations of receptor tyrosine kinases and Ras genes in childhood core-binding factor acute myeloid leukemia and a comparative analysis on paired diagnosis and relapse samples. Leukemia 22 (2): 303-7, 2008.
- Goemans BF, Zwaan CM, Miller M, et al.: Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia 19 (9): 1536-42, 2005.
- Pollard JA, Alonzo TA, Gerbing RB, et al.: Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood 115 (12): 2372-9, 2010.
- Shimada A, Taki T, Tabuchi K, et al.: KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t(8;21): a study of the Japanese Childhood AML Cooperative Study Group. Blood 107 (5): 1806-9, 2006.
- Manara E, Bisio V, Masetti R, et al.: Core-binding factor acute myeloid leukemia in pediatric patients enrolled in the AIEOP AML 2002/01 trial: screening and prognostic impact of c-KIT mutations. Leukemia 28 (5): 1132-4, 2014.
- Chen X, Dou H, Wang X, et al.: KIT mutations correlate with adverse survival in children with core-binding factor acute myeloid leukemia. Leuk Lymphoma 59 (4): 829-836, 2018.
- Tokumasu M, Murata C, Shimada A, et al.: Adverse prognostic impact of KIT mutations in childhood CBF-AML: the results of the Japanese Pediatric Leukemia/Lymphoma Study Group AML-05 trial. Leukemia 29 (12): 2438-41, 2015.
- Jahn N, Agrawal M, Bullinger L, et al.: Incidence and prognostic impact of ASXL2 mutations in adult acute myeloid leukemia patients with t(8;21)(q22;q22): a study of the German-Austrian AML Study Group. Leukemia 31 (4): 1012-1015, 2017.
- Yamato G, Shiba N, Yoshida K, et al.: ASXL2 mutations are frequently found in pediatric AML patients with t(8;21)/ RUNX1-RUNX1T1 and associated with a better prognosis. Genes Chromosomes Cancer 56 (5): 382-393, 2017.
- Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34. Also available online. Last accessed August 11, 2022.
- Sanz MA, Grimwade D, Tallman MS, et al.: Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 113 (9): 1875-91, 2009.
- Licht JD, Chomienne C, Goy A, et al.: Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood 85 (4): 1083-94, 1995.
- Yan W, Zhang G: Molecular Characteristics and Clinical Significance of 12 Fusion Genes in Acute Promyelocytic Leukemia: A Systematic Review. Acta Haematol 136 (1): 1-15, 2016.
- Döhner K, Schlenk RF, Habdank M, et al.: Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood 106 (12): 3740-6, 2005.
- Verhaak RG, Goudswaard CS, van Putten W, et al.: Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood 106 (12): 3747-54, 2005.
- Schnittger S, Schoch C, Kern W, et al.: Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 106 (12): 3733-9, 2005.
- Falini B, Mecucci C, Tiacci E, et al.: Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 352 (3): 254-66, 2005.
- Schlenk RF, Döhner K, Krauter J, et al.: Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 358 (18): 1909-18, 2008.
- Gale RE, Green C, Allen C, et al.: The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 111 (5): 2776-84, 2008.
- Cazzaniga G, Dell'Oro MG, Mecucci C, et al.: Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood 106 (4): 1419-22, 2005.
- Balgobind BV, Hollink IH, Arentsen-Peters ST, et al.: Integrative analysis of type-I and type-II aberrations underscores the genetic heterogeneity of pediatric acute myeloid leukemia. Haematologica 96 (10): 1478-87, 2011.
- Staffas A, Kanduri M, Hovland R, et al.: Presence of FLT3-ITD and high BAALC expression are independent prognostic markers in childhood acute myeloid leukemia. Blood 118 (22): 5905-13, 2011.
- Tawana K, Wang J, Renneville A, et al.: Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood 126 (10): 1214-23, 2015.
- Tarlock K, Lamble AJ, Wang YC, et al.: CEBPA-bZip mutations are associated with favorable prognosis in de novo AML: a report from the Children's Oncology Group. Blood 138 (13): 1137-1147, 2021.
- Marcucci G, Maharry K, Radmacher MD, et al.: Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. J Clin Oncol 26 (31): 5078-87, 2008.
- Wouters BJ, Löwenberg B, Erpelinck-Verschueren CA, et al.: Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 113 (13): 3088-91, 2009.
- Dufour A, Schneider F, Metzeler KH, et al.: Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol 28 (4): 570-7, 2010.
- Taskesen E, Bullinger L, Corbacioglu A, et al.: Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood 117 (8): 2469-75, 2011.
- Fasan A, Haferlach C, Alpermann T, et al.: The role of different genetic subtypes of CEBPA mutated AML. Leukemia 28 (4): 794-803, 2014.
- Taube F, Georgi JA, Kramer M, et al.: CEBPA mutations in 4708 patients with acute myeloid leukemia: differential impact of bZIP and TAD mutations on outcome. Blood 139 (1): 87-103, 2022.
- Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, et al.: Characterization of CEBPA mutations and promoter hypermethylation in pediatric acute myeloid leukemia. Haematologica 96 (3): 384-92, 2011.
- Tarlock K, Alonzo T, Wang YC, et al.: Prognostic impact of CSF3R mutations in favorable risk childhood acute myeloid leukemia. Blood 135 (18): 1603-1606, 2020.
- Groet J, McElwaine S, Spinelli M, et al.: Acquired mutations in GATA1 in neonates with Down's syndrome with transient myeloid disorder. Lancet 361 (9369): 1617-20, 2003.
- Hitzler JK, Cheung J, Li Y, et al.: GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 101 (11): 4301-4, 2003.
- Rainis L, Bercovich D, Strehl S, et al.: Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood 102 (3): 981-6, 2003.
- Wechsler J, Greene M, McDevitt MA, et al.: Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 32 (1): 148-52, 2002.
- de Rooij JD, Branstetter C, Ma J, et al.: Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet 49 (3): 451-456, 2017.
- Ge Y, Stout ML, Tatman DA, et al.: GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst 97 (3): 226-31, 2005.
- Mrózek K, Heerema NA, Bloomfield CD: Cytogenetics in acute leukemia. Blood Rev 18 (2): 115-36, 2004.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Outcome of pediatric patients with acute myeloid leukemia (AML) and -5/5q- abnormalities from five pediatric AML treatment protocols: a report from the Children's Oncology Group. Pediatr Blood Cancer 60 (12): 2073-8, 2013.
- Stevens RF, Hann IM, Wheatley K, et al.: Marked improvements in outcome with chemotherapy alone in paediatric acute myeloid leukemia: results of the United Kingdom Medical Research Council's 10th AML trial. MRC Childhood Leukaemia Working Party. Br J Haematol 101 (1): 130-40, 1998.
- Hasle H, Alonzo TA, Auvrignon A, et al.: Monosomy 7 and deletion 7q in children and adolescents with acute myeloid leukemia: an international retrospective study. Blood 109 (11): 4641-7, 2007.
- Rasche M, von Neuhoff C, Dworzak M, et al.: Genotype-outcome correlations in pediatric AML: the impact of a monosomal karyotype in trial AML-BFM 2004. Leukemia 31 (12): 2807-2814, 2017.
- Blink M, Zimmermann M, von Neuhoff C, et al.: Normal karyotype is a poor prognostic factor in myeloid leukemia of Down syndrome: a retrospective, international study. Haematologica 99 (2): 299-307, 2014.
- Hammer ASB, Juul-Dam KL, Sandahl JD, et al.: Hypodiploidy has unfavorable impact on survival in pediatric acute myeloid leukemia: an I-BFM Study Group collaboration. Blood Adv 7 (6): 1045-1055, 2023.
- Gröschel S, Sanders MA, Hoogenboezem R, et al.: A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157 (2): 369-81, 2014.
- Yamazaki H, Suzuki M, Otsuki A, et al.: A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 25 (4): 415-27, 2014.
- Lugthart S, Gröschel S, Beverloo HB, et al.: Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol 28 (24): 3890-8, 2010.
- Balgobind BV, Lugthart S, Hollink IH, et al.: EVI1 overexpression in distinct subtypes of pediatric acute myeloid leukemia. Leukemia 24 (5): 942-9, 2010.
- Schnittger S, Schoch C, Dugas M, et al.: Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 100 (1): 59-66, 2002.
- Thiede C, Steudel C, Mohr B, et al.: Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 99 (12): 4326-35, 2002.
- Iwai T, Yokota S, Nakao M, et al.: Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. The Children's Cancer and Leukemia Study Group, Japan. Leukemia 13 (1): 38-43, 1999.
- Meshinchi S, Stirewalt DL, Alonzo TA, et al.: Activating mutations of RTK/ras signal transduction pathway in pediatric acute myeloid leukemia. Blood 102 (4): 1474-9, 2003.
- Zwaan CM, Meshinchi S, Radich JP, et al.: FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood 102 (7): 2387-94, 2003.
- Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, et al.: NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 118 (13): 3645-56, 2011.
- Ostronoff F, Othus M, Gerbing RB, et al.: NUP98/NSD1 and FLT3/ITD coexpression is more prevalent in younger AML patients and leads to induction failure: a COG and SWOG report. Blood 124 (15): 2400-7, 2014.
- Arrigoni P, Beretta C, Silvestri D, et al.: FLT3 internal tandem duplication in childhood acute myeloid leukaemia: association with hyperleucocytosis in acute promyelocytic leukaemia. Br J Haematol 120 (1): 89-92, 2003.
- Kutny MA, Moser BK, Laumann K, et al.: FLT3 mutation status is a predictor of early death in pediatric acute promyelocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 59 (4): 662-7, 2012.
- Gale RE, Hills R, Pizzey AR, et al.: Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood 106 (12): 3768-76, 2005.
- Tallman MS, Kim HT, Montesinos P, et al.: Does microgranular variant morphology of acute promyelocytic leukemia independently predict a less favorable outcome compared with classical M3 APL? A joint study of the North American Intergroup and the PETHEMA Group. Blood 116 (25): 5650-9, 2010.
- Sung L, Aplenc R, Alonzo TA, et al.: Predictors and short-term outcomes of hyperleukocytosis in children with acute myeloid leukemia: a report from the Children's Oncology Group. Haematologica 97 (11): 1770-3, 2012.
- Callens C, Chevret S, Cayuela JM, et al.: Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia 19 (7): 1153-60, 2005.
- Schnittger S, Bacher U, Haferlach C, et al.: Clinical impact of FLT3 mutation load in acute promyelocytic leukemia with t(15;17)/PML-RARA. Haematologica 96 (12): 1799-807, 2011.
- Breccia M, Loglisci G, Loglisci MG, et al.: FLT3-ITD confers poor prognosis in patients with acute promyelocytic leukemia treated with AIDA protocols: long-term follow-up analysis. Haematologica 98 (12): e161-3, 2013.
- Poiré X, Moser BK, Gallagher RE, et al.: Arsenic trioxide in front-line therapy of acute promyelocytic leukemia (C9710): prognostic significance of FLT3 mutations and complex karyotype. Leuk Lymphoma 55 (7): 1523-32, 2014.
- Pui CH, Relling MV, Rivera GK, et al.: Epipodophyllotoxin-related acute myeloid leukemia: a study of 35 cases. Leukemia 9 (12): 1990-6, 1995.
- Inaba H, Zhou Y, Abla O, et al.: Heterogeneous cytogenetic subgroups and outcomes in childhood acute megakaryoblastic leukemia: a retrospective international study. Blood 126 (13): 1575-84, 2015.
- Balgobind BV, Raimondi SC, Harbott J, et al.: Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood 114 (12): 2489-96, 2009.
- Swansbury GJ, Slater R, Bain BJ, et al.: Hematological malignancies with t(9;11)(p21-22;q23)--a laboratory and clinical study of 125 cases. European 11q23 Workshop participants. Leukemia 12 (5): 792-800, 1998.
- Pollard JA, Guest E, Alonzo TA, et al.: Gemtuzumab Ozogamicin Improves Event-Free Survival and Reduces Relapse in Pediatric KMT2A-Rearranged AML: Results From the Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 39 (28): 3149-3160, 2021.
- Coenen EA, Raimondi SC, Harbott J, et al.: Prognostic significance of additional cytogenetic aberrations in 733 de novo pediatric 11q23/MLL-rearranged AML patients: results of an international study. Blood 117 (26): 7102-11, 2011.
- Ageberg M, Drott K, Olofsson T, et al.: Identification of a novel and myeloid specific role of the leukemia-associated fusion protein DEK-NUP214 leading to increased protein synthesis. Genes Chromosomes Cancer 47 (4): 276-87, 2008.
- Shiba N, Ichikawa H, Taki T, et al.: NUP98-NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes Chromosomes Cancer 52 (7): 683-93, 2013.
- Slovak ML, Gundacker H, Bloomfield CD, et al.: A retrospective study of 69 patients with t(6;9)(p23;q34) AML emphasizes the need for a prospective, multicenter initiative for rare 'poor prognosis' myeloid malignancies. Leukemia 20 (7): 1295-7, 2006.
- Alsabeh R, Brynes RK, Slovak ML, et al.: Acute myeloid leukemia with t(6;9) (p23;q34): association with myelodysplasia, basophilia, and initial CD34 negative immunophenotype. Am J Clin Pathol 107 (4): 430-7, 1997.
- Sandahl JD, Coenen EA, Forestier E, et al.: t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: an international study of 62 patients. Haematologica 99 (5): 865-72, 2014.
- Tarlock K, Alonzo TA, Moraleda PP, et al.: Acute myeloid leukaemia (AML) with t(6;9)(p23;q34) is associated with poor outcome in childhood AML regardless of FLT3-ITD status: a report from the Children's Oncology Group. Br J Haematol 166 (2): 254-9, 2014.
- Gruber TA, Larson Gedman A, Zhang J, et al.: An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 22 (5): 683-97, 2012.
- Thiollier C, Lopez CK, Gerby B, et al.: Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J Exp Med 209 (11): 2017-31, 2012.
- de Rooij JD, Hollink IH, Arentsen-Peters ST, et al.: NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 27 (12): 2280-8, 2013.
- Masetti R, Pigazzi M, Togni M, et al.: CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 121 (17): 3469-72, 2013.
- Masetti R, Rondelli R, Fagioli F, et al.: Infants with acute myeloid leukemia treated according to the Associazione Italiana di Ematologia e Oncologia Pediatrica 2002/01 protocol have an outcome comparable to that of older children. Haematologica 99 (8): e127-9, 2014.
- de Rooij JD, Masetti R, van den Heuvel-Eibrink MM, et al.: Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood 127 (26): 3424-30, 2016.
- Hara Y, Shiba N, Ohki K, et al.: Prognostic impact of specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down syndrome. Genes Chromosomes Cancer 56 (5): 394-404, 2017.
- Smith JL, Ries RE, Hylkema T, et al.: Comprehensive Transcriptome Profiling of Cryptic CBFA2T3-GLIS2 Fusion-Positive AML Defines Novel Therapeutic Options: A COG and TARGET Pediatric AML Study. Clin Cancer Res 26 (3): 726-737, 2020.
- Eidenschink Brodersen L, Alonzo TA, Menssen AJ, et al.: A recurrent immunophenotype at diagnosis independently identifies high-risk pediatric acute myeloid leukemia: a report from Children's Oncology Group. Leukemia 30 (10): 2077-2080, 2016.
- Pardo LM, Voigt AP, Alonzo TA, et al.: Deciphering the Significance of CD56 Expression in Pediatric Acute Myeloid Leukemia: A Report from the Children's Oncology Group. Cytometry B Clin Cytom 98 (1): 52-56, 2020.
- Noort S, Wander P, Alonzo TA, et al.: The clinical and biological characteristics of NUP98-KDM5A in pediatric acute myeloid leukemia. Haematologica 106 (2): 630-634, 2021.
- Lion T, Haas OA: Acute megakaryocytic leukemia with the t(1;22)(p13;q13). Leuk Lymphoma 11 (1-2): 15-20, 1993.
- Duchayne E, Fenneteau O, Pages MP, et al.: Acute megakaryoblastic leukaemia: a national clinical and biological study of 53 adult and childhood cases by the Groupe Français d'Hématologie Cellulaire (GFHC). Leuk Lymphoma 44 (1): 49-58, 2003.
- Ma Z, Morris SW, Valentine V, et al.: Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat Genet 28 (3): 220-1, 2001.
- Mercher T, Coniat MB, Monni R, et al.: Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22) translocation of acute megakaryocytic leukemia. Proc Natl Acad Sci U S A 98 (10): 5776-9, 2001.
- Bernstein J, Dastugue N, Haas OA, et al.: Nineteen cases of the t(1;22)(p13;q13) acute megakaryblastic leukaemia of infants/children and a review of 39 cases: report from a t(1;22) study group. Leukemia 14 (1): 216-8, 2000.
- Coenen EA, Zwaan CM, Reinhardt D, et al.: Pediatric acute myeloid leukemia with t(8;16)(p11;p13), a distinct clinical and biological entity: a collaborative study by the International-Berlin-Frankfurt-Munster AML-study group. Blood 122 (15): 2704-13, 2013.
- Wong KF, Yuen HL, Siu LL, et al.: t(8;16)(p11;p13) predisposes to a transient but potentially recurring neonatal leukemia. Hum Pathol 39 (11): 1702-7, 2008.
- Wu X, Sulavik D, Roulston D, et al.: Spontaneous remission of congenital acute myeloid leukemia with t(8;16)(p11;13). Pediatr Blood Cancer 56 (2): 331-2, 2011.
- Terui K, Sato T, Sasaki S, et al.: Two novel variants of MOZ-CBP fusion transcripts in spontaneously remitted infant leukemia with t(1;16;8)(p13;p13;p11), a new variant of t(8;16)(p11;p13). Haematologica 93 (10): 1591-3, 2008.
- von Bergh AR, van Drunen E, van Wering ER, et al.: High incidence of t(7;12)(q36;p13) in infant AML but not in infant ALL, with a dismal outcome and ectopic expression of HLXB9. Genes Chromosomes Cancer 45 (8): 731-9, 2006.
- Tosi S, Harbott J, Teigler-Schlegel A, et al.: t(7;12)(q36;p13), a new recurrent translocation involving ETV6 in infant leukemia. Genes Chromosomes Cancer 29 (4): 325-32, 2000.
- Slater RM, von Drunen E, Kroes WG, et al.: t(7;12)(q36;p13) and t(7;12)(q32;p13)--translocations involving ETV6 in children 18 months of age or younger with myeloid disorders. Leukemia 15 (6): 915-20, 2001.
- Park J, Kim M, Lim J, et al.: Three-way complex translocations in infant acute myeloid leukemia with t(7;12)(q36;p13): the incidence and correlation of a HLXB9 overexpression. Cancer Genet Cytogenet 191 (2): 102-5, 2009.
- Takeda A, Yaseen NR: Nucleoporins and nucleocytoplasmic transport in hematologic malignancies. Semin Cancer Biol 27: 3-10, 2014.
- Panarello C, Rosanda C, Morerio C: Cryptic translocation t(5;11)(q35;p15.5) with involvement of the NSD1 and NUP98 genes without 5q deletion in childhood acute myeloid leukemia. Genes Chromosomes Cancer 35 (3): 277-81, 2002.
- Cerveira N, Correia C, Dória S, et al.: Frequency of NUP98-NSD1 fusion transcript in childhood acute myeloid leukaemia. Leukemia 17 (11): 2244-7, 2003.
- McNeer NA, Philip J, Geiger H, et al.: Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia 33 (8): 1934-1943, 2019.
- van Zutven LJ, Onen E, Velthuizen SC, et al.: Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes Chromosomes Cancer 45 (5): 437-46, 2006.
- Yamato G, Shiba N, Yoshida K, et al.: RUNX1 mutations in pediatric acute myeloid leukemia are associated with distinct genetic features and an inferior prognosis. Blood 131 (20): 2266-2270, 2018.
- Berman JN, Gerbing RB, Alonzo TA, et al.: Prevalence and clinical implications of NRAS mutations in childhood AML: a report from the Children's Oncology Group. Leukemia 25 (6): 1039-42, 2011.
- Kühn MW, Radtke I, Bullinger L, et al.: High-resolution genomic profiling of adult and pediatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood 119 (10): e67-75, 2012.
- Schnittger S, Kohl TM, Haferlach T, et al.: KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood 107 (5): 1791-9, 2006.
- Paschka P, Marcucci G, Ruppert AS, et al.: Wilms' tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol 26 (28): 4595-602, 2008.
- Virappane P, Gale R, Hills R, et al.: Mutation of the Wilms' tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the United Kingdom Medical Research Council Adult Leukaemia Working Party. J Clin Oncol 26 (33): 5429-35, 2008.
- Gaidzik VI, Schlenk RF, Moschny S, et al.: Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: a study of the German-Austrian AML Study Group. Blood 113 (19): 4505-11, 2009.
- Renneville A, Boissel N, Zurawski V, et al.: Wilms tumor 1 gene mutations are associated with a higher risk of recurrence in young adults with acute myeloid leukemia: a study from the Acute Leukemia French Association. Cancer 115 (16): 3719-27, 2009.
- Ho PA, Zeng R, Alonzo TA, et al.: Prevalence and prognostic implications of WT1 mutations in pediatric acute myeloid leukemia (AML): a report from the Children's Oncology Group. Blood 116 (5): 702-10, 2010.
- Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M, et al.: Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood 113 (23): 5951-60, 2009.
- Ley TJ, Ding L, Walter MJ, et al.: DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363 (25): 2424-33, 2010.
- Yan XJ, Xu J, Gu ZH, et al.: Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet 43 (4): 309-15, 2011.
- Thol F, Damm F, Lüdeking A, et al.: Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol 29 (21): 2889-96, 2011.
- Ho PA, Kutny MA, Alonzo TA, et al.: Leukemic mutations in the methylation-associated genes DNMT3A and IDH2 are rare events in pediatric AML: a report from the Children's Oncology Group. Pediatr Blood Cancer 57 (2): 204-9, 2011.
- Green CL, Evans CM, Hills RK, et al.: The prognostic significance of IDH1 mutations in younger adult patients with acute myeloid leukemia is dependent on FLT3/ITD status. Blood 116 (15): 2779-82, 2010.
- Paschka P, Schlenk RF, Gaidzik VI, et al.: IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 28 (22): 3636-43, 2010.
- Abbas S, Lugthart S, Kavelaars FG, et al.: Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 116 (12): 2122-6, 2010.
- Marcucci G, Maharry K, Wu YZ, et al.: IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 28 (14): 2348-55, 2010.
- Wagner K, Damm F, Göhring G, et al.: Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol 28 (14): 2356-64, 2010.
- Figueroa ME, Abdel-Wahab O, Lu C, et al.: Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18 (6): 553-67, 2010.
- Ward PS, Patel J, Wise DR, et al.: The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17 (3): 225-34, 2010.
- Dang L, White DW, Gross S, et al.: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462 (7274): 739-44, 2009.
- Damm F, Thol F, Hollink I, et al.: Prevalence and prognostic value of IDH1 and IDH2 mutations in childhood AML: a study of the AML-BFM and DCOG study groups. Leukemia 25 (11): 1704-10, 2011.
- Oki K, Takita J, Hiwatari M, et al.: IDH1 and IDH2 mutations are rare in pediatric myeloid malignancies. Leukemia 25 (2): 382-4, 2011.
- Pigazzi M, Ferrari G, Masetti R, et al.: Low prevalence of IDH1 gene mutation in childhood AML in Italy. Leukemia 25 (1): 173-4, 2011.
- Ho PA, Alonzo TA, Kopecky KJ, et al.: Molecular alterations of the IDH1 gene in AML: a Children's Oncology Group and Southwest Oncology Group study. Leukemia 24 (5): 909-13, 2010.
- Andersson AK, Miller DW, Lynch JA, et al.: IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia 25 (10): 1570-7, 2011.
- Maxson JE, Ries RE, Wang YC, et al.: CSF3R mutations have a high degree of overlap with CEBPA mutations in pediatric AML. Blood 127 (24): 3094-8, 2016.
- Germeshausen M, Kratz CP, Ballmaier M, et al.: RAS and CSF3R mutations in severe congenital neutropenia. Blood 114 (16): 3504-5, 2009.
- Skokowa J, Steinemann D, Katsman-Kuipers JE, et al.: Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: a unique pathway in myeloid leukemogenesis. Blood 123 (14): 2229-37, 2014.
Treatment Option Overview for Childhood AML
Leukemia is considered to be disseminated in the hematopoietic system at diagnosis, even in children with acute myeloid leukemia (AML) who present with isolated chloromas (also called granulocytic or myeloid sarcomas). If these children do not receive systemic chemotherapy, they invariably develop AML in months or years. AML may invade nonhematopoietic (extramedullary) tissue such as meninges, brain parenchyma, testes or ovaries, or skin (leukemia cutis). Extramedullary leukemia is more common in infants than in older children with AML.[1]
Childhood AML is diagnosed when bone marrow has 20% or greater blasts. The blasts have the morphological and histochemical characteristics of one of the French-American-British (FAB) subtypes of AML. It can also be diagnosed by biopsy of a chloroma. For treatment purposes, patients with clonal cytogenetic abnormalities typically associated with AML, such as t(8;21)(RUNX1::RUNX1T1 gene fusions), inv(16)(CBFB::MYH11 gene fusions), t(9;11)(MLLT3::KMT2A gene fusions) or t(15;17)(PML::RARA gene fusions) and who have less than 20% bone marrow blasts, are considered to have AML rather than a myelodysplastic syndrome.[2]
Complete remission (CR) has traditionally been defined in the United States using morphological criteria such as the following:
- Peripheral blood counts (white blood cell [WBC] count, differential [absolute neutrophil count >1,000/μL], and platelet count >100,000/μL) rising toward normal.
- Mildly hypocellular to normal cellular marrow with fewer than 5% blasts.
- No clinical signs or symptoms of the disease, including in the central nervous system (CNS) or at other extramedullary sites.[3]
Alternative definitions of remission using morphology are used in AML because of the prolonged myelosuppression caused by intensive chemotherapy and include CR with incomplete platelet recovery (CRp) and CR with incomplete marrow recovery (typically absolute neutrophil count) (CRi). Whereas the use of CRp provides a clinically meaningful response, the traditional CR definition remains the gold standard because patients in CR were found to be more likely to survive longer than those in CRp.[4]
Achieving a hypoplastic bone marrow (using morphology) is usually the first step in obtaining remission in AML with the exception of the M3 subtype (acute promyelocytic leukemia [APL]); a hypoplastic marrow phase is often not necessary before the achievement of remission in APL. Additionally, early recovery marrows in any of the subtypes of AML may be difficult to distinguish from persistent leukemia, although the application of flow cytometric immunophenotyping and cytogenetic/molecular testing have made this less problematic. Correlation with blood cell counts and clinical status is imperative in passing final judgment on the results of early bone marrow findings in AML.[5] If the findings are in doubt, the bone marrow aspirate should be repeated in 1 to 2 weeks.[1]
In addition to morphology, more precise methodology (e.g., multiparameter flow cytometry or quantitative reverse transcriptase–polymerase chain reaction [RT-PCR]) is used to assess response and has been shown to be of greater prognostic significance than morphology. For more information about these methodologies, see the Prognostic Factors in Childhood AML section.
Treatment Approach
The mainstay of the therapeutic approach is systemically administered combination chemotherapy. Approaches involving risk-group stratification and biologically targeted therapies are being tested to improve antileukemic treatment while sparing normal tissue. Optimal treatment of AML requires control of bone marrow and systemic disease. Treatment of the CNS, usually with intrathecal medication, is a component of most pediatric AML protocols but has not yet been shown to contribute directly to an improvement in survival. CNS irradiation is not necessary in patients, either as prophylaxis or for those presenting with cerebrospinal fluid leukemia that clears with intrathecal and systemic chemotherapy.
Treatment is ordinarily divided into the following two phases:
- Induction (to induce remission).
- Postremission consolidation/intensification (to reduce the risk of relapse).
Postremission therapy may consist of varying numbers of courses of intensive chemotherapy and/or allogeneic hematopoietic stem cell transplantation (HSCT). For example, ongoing trials of the Children's Oncology Group (COG) and the United Kingdom Medical Research Council (MRC) use similar chemotherapy regimens consisting of two courses of induction chemotherapy followed by two to three additional courses of intensification chemotherapy.[6,7,8]
Maintenance therapy is not part of most pediatric AML protocols because two randomized clinical trials failed to show a benefit for maintenance therapy when given after modern intensive chemotherapy.[9,10] The exception to this generalization is made for APL, because maintenance therapy was shown to improve event-free survival (EFS) and overall survival (OS) when tretinoin was combined with chemotherapy.[11] Some studies of adult APL patients, including studies incorporating arsenic trioxide treatment, have shown no benefit to maintenance.[12,13]
Attention to both acute and long-term complications is critical in children with AML. Modern AML treatment approaches are usually associated with severe, protracted myelosuppression with related complications. Children with AML should receive care under the direction of pediatric oncologists in cancer centers or hospitals with appropriate supportive care facilities (e.g., specialized blood products; pediatric intensive care; provision of emotional and developmental support). With improved supportive care, toxic death constitutes a smaller proportion of initial therapy failures than in the past.[6] Two COG trials reported an 11% to 13% incidence of remission failure, mainly because of resistant disease. Only 2% to 3% resulted from toxic death during the two induction courses.[8,14]
Children treated for AML are living longer and require close monitoring for cancer therapy side effects that may persist or develop months or years after treatment. The high cumulative doses of anthracyclines require long-term monitoring of cardiac function. The use of some modalities have declined, including total-body irradiation with HSCT because of its increased risk of growth failure, gonadal and thyroid dysfunction, cataract formation, and second malignancies.[15] For more information, see the Survivorship and Adverse Late Sequelae section or Late Effects of Treatment for Childhood Cancer.
Prognostic Factors in Childhood AML
Prognostic factors in childhood AML can be categorized as follows:
- Host risk factors.
- Leukemia risk factors.
- Therapeutic response risk factors.
Host risk factors
- Age: Several reports have identified older age as an adverse prognostic factor.[7,16,17,18,19,20] The age effect is not large with regard to OS, but in general, the adverse outcomes seen in adolescents compared with younger children appear to be primarily caused by increases in toxic mortality.[21] In the COG AAML1031 (NCT01371981) trial, age older than 11 years was an independent predictor of more favorable EFS on multivariable analysis.[22]
While outcome for infants with ALL remains inferior to that of older children, outcome for infants with AML is similar to that of older children when they are treated with standard AML regimens.[16,23,24,25] Infants have been reported to have a 5-year survival rate of 60% to 70%, although with increased treatment-associated toxicity, particularly during induction.[16,23,24,25,26]
- Race and ethnicity: In both the Children's Cancer Group (CCG) CCG-2891 and COG-2961 (NCT00002798) studies, White children had higher OS rates than did Black and Hispanic children.[18,27,28] Black children also experienced lower survival rates than White children in St. Jude Children's Research Hospital AML clinical trials.[29]
- Down syndrome: For children with Down syndrome who develop AML, survival is generally favorable when diagnosed at a young age.[30,31,32] The prognosis is particularly good (EFS rate exceeding 80%) for children younger than 4 years at diagnosis, the age group that accounts for the vast majority of Down syndrome patients with AML, whereas those older than 4 years have similar outcomes to those without Down syndrome.[32,33,34,35,36]
- Body mass index: Obesity (body mass index more than 95th percentile for age) is predictive of inferior survival.[18,37] Inferior survival was attributable to early treatment-related mortality that was primarily caused by infectious complications.[37,38]
Leukemia risk factors
- White blood cell (WBC) count: WBC count at diagnosis has been consistently noted to be inversely related to survival.[7,22,39,40] Patients with high presenting leukocyte counts have a higher risk of developing pulmonary and CNS complications and, historically, have a higher risk of induction death.[41]
In patients with APL, WBC at initial diagnosis alone is used to distinguish standard-risk and high-risk APL. A WBC count of 10,000 cells/μL or more denotes high risk, and these patients have an increased risk of both early death and relapse.[42] However, more recent regimens that include arsenic trioxide result in low relapse rates that are not significantly different between the high-risk and low-risk groups.[43]
- FAB subtype: Associations between FAB subtype and prognosis have been more variable.
- M3 subtype. The M3 (APL) subtype has a favorable outcome in studies using tretinoin in combination with chemotherapy and arsenic trioxide consolidation.[42,43,44,45]
- M6 subtype. In the 2016 WHO classification system, the M6 subtype was limited to pure erythroid leukemia. The combined COG AAML0531 and AAML1031 studies demonstrated that it is a rare subtype (5 of 1,934 cases; 0.2%), occurred in younger patients (median age, 2.3 years), and was associated with a poor outcome (5-year EFS and OS rates, 20% ± 36%).[46]
- M7 subtype. Some studies have indicated a relatively poor outcome for M7 (megakaryocytic leukemia) in patients without Down syndrome,[30] although reports suggest an intermediate prognosis for this group of patients when contemporary treatment approaches are used.[6,47,48]
In a retrospective study of non–Down syndrome M7 patients with samples available for molecular analysis, the presence of specific genetic abnormalities (CBFA2T3::GLIS2 gene fusions [cryptic inv(16)(p13q24)], NUP98::KDM5A gene fusions, t(11;12)(p15;p13), KMT2A [MLL] rearrangements, monosomy 7) was associated with a significantly worse outcome than for other M7 patients.[49,50] In contrast, 10% of non–Down syndrome AMKL patients with GATA1 mutations had favorable outcomes when there were no prognostically unfavorable gene fusions present. This was also true for patients with HOX rearrangements.[50]
- M0 subtype. The M0, or minimally differentiated subtype, has been associated with a poor outcome.[51]
- CNS disease: CNS involvement at diagnosis is categorized on the basis of the presence or absence of blasts in cerebrospinal fluid (CSF). European cooperative groups have applied acute lymphoblastic leukemia–inspired definitions to AML, as follows:
- CNS1: CSF negative for blasts on cytospin, regardless of CSF WBC count.
- CNS2 is divided into the following three groups and defined as follows:
- CNS2a: CSF with fewer than 5 WBC/μL and cytospin positive for blasts in an atraumatic tap (<10 red blood cells [RBC]/μL).
- CNS2b: CSF with fewer than 5 WBC/μL and cytospin positive for blasts in a traumatic tap (≥10 RBC/μL).
- CNS2c: CSF with 5 or more WBC/μL and cytospin positive for blasts in a traumatic tap (≥10 RBC/μL) in which the WBC/RBC ratio in the CSF is less than twice that in the peripheral blood.
- CNS3 includes the following three subgroups and is defined as follows:
- CNS3a: CSF with 5 or more WBC/μL and cytospin positive for blasts in an atraumatic tap (<10 RBC/μL).
- CNS3b: CSF with 5 or more WBC/μL and cytospin positive for blasts in a traumatic tap (≥10 RBC/μL) in which the WBC/RBC ratio in the CSF is more than or equal to twice the ratio in the peripheral blood.
- CNS3c: Clinical signs of CNS leukemia (e.g., cranial nerve palsy, brain/eye involvement, or radiographic evidence of an intracranial, intradural chloroma).
COG trials (including AAML03P1 [NCT00070174], AAML0531 [NCT00372593], and AAML1031 [NCT01371981]) used a modified version of the CNS disease definitions, in which patients were dichotomously classified for treatment purposes as CNS positive or negative. The CNS-positive group included all patients with blasts on cytospin (regardless of CSF WBC) unless there were more than 100 RBC/μL in the CSF. Patients with 100 RBC/μL in the CSF were CNS positive only if the WBC/RBC ratio in the CSF was greater than or equal to twice the ratio in the peripheral blood. CNS outcomes on COG studies were analyzed utilizing the more traditional CNS1/2/3 definitions.[52]
CNS2 disease has been observed in approximately 13% to 16% of children with AML, and CNS3 disease has been observed in approximately 11% to 17% of children with AML.[52,53] Studies have variably shown that patients with CNS2/CNS3 were younger, more often had hyperleukocytosis, and had higher incidences of t(9;11), t(8;21), or inv(16).[52,53]
While CNS involvement (CNS2 or CNS3) at diagnosis has not been shown to be correlated with OS in most studies, a COG analysis of children with AML enrolled from 2003 to 2010 on two consecutive and identical backbone trials found that CNS involvement, especially CNS3 status, was associated with inferior outcomes, including complete remission rate, EFS, disease-free survival, and an increased risk of relapse involving the CNS.[52] Another trial showed it to be associated with an increased risk of isolated CNS relapse.[54] Finally, the COG study did not find an adverse impact of traumatic lumbar punctures at diagnosis upon eventual outcome.[52]
- Cytogenetic and molecular characteristics: Cytogenetic and molecular characteristics are also associated with prognosis. For detailed information, see the Molecular Evaluation section. Cytogenetic and molecular characteristics that are currently used in the COG clinical trials for treatment assignment include the following:
- Favorable: inv(16)/t(16;16), t(8;21), t(15;17), biallelic CEBPA mutations, and NPM1 mutations.
- Unfavorable: monosomy 7, monosomy 5/del(5q), 3q abnormalities, FLT3 internal tandem duplications (ITD) with high-allelic ratio, KMT2A gene fusions with the following partners: t(4;11)(q21;q23), t(6;11)(q27;q23), t(10;11)(p11.2;q23), t(10;11)(p12;q23), or t(11;19)(q23;p13.3), NUP98 (11p15.5), 12p (ETV6 rearrangement or loss), t(16;21)(p11;q22)(FUS::ERG gene fusions), CBFA2T3::GLIS2 gene fusions, KAT6A (8p11.21) (if over 90 days of age), and non–KMT2A::MLLT10 gene fusions.[55,56]
- Immunophenotype:
- A distinctive immunophenotype (initially reported as the RAM phenotype), with high CD56 levels, dim or negative expression of CD45 and CD38, and a lack of HLA-DR expression was associated with a poor prognosis (5-year EFS rate of approximately 20%).[57,58] Most patients with the RAM phenotype have the CBFA2T3::GLIS2 fusion.[58,59]
- High CD123 expression (quartile 4 vs. quartiles 1–3), in Cox multivariable regression, was shown to be an independent adverse prognostic risk factor for OS, EFS, and relapse risk (RR), although it did not impact remission success. High CD123 expression occurred more frequently in patients with many high-risk cytogenetic and molecular characteristics. High CD123 expression also adversely impacted OS and EFS, but not RR. In patients with low-risk cytogenetic and molecular characteristics, those with high CD123 expression (quartile 4) had significantly worse OS, EFS, and RR.[60]
Therapeutic response risk factors
- Response to therapy/minimal residual disease (MRD): Early response to therapy, generally measured after the first course of induction therapy, is predictive of outcome and can be assessed by standard morphological examination of bone marrow,[39,61] cytogenetic analysis, fluorescence in situ hybridization, or more sophisticated techniques to identify MRD (e.g., multiparameter flow cytometry, quantitative RT-PCR).[62,63,64] Multiple groups have shown that the level of MRD after one course of induction therapy is an independent predictor of prognosis.[62,63,64,65,66,67]
Molecular approaches to assessing MRD in AML (e.g., using quantitative RT-PCR) have been challenging to apply because of the genomic heterogeneity of pediatric AML and the instability of some genomic alterations. Quantitative RT-PCR detection of RUNX1::RUNX1T1 fusion transcripts can effectively predict higher risk of relapse for patients in clinical remission.[68,69,70] Other molecular alterations such as NPM1 mutations [71] and CBFB::MYH11 fusion transcripts [72] have also been successfully employed as leukemia-specific molecular markers in MRD assays; for these alterations, the level of MRD has shown prognostic significance. The presence of FLT3 ITD has been shown to be discordant between diagnosis and relapse, although when its presence persists (usually associated with a high-allelic ratio at diagnosis), it can be useful in detecting residual leukemia.[73]
For APL, MRD detection at the end of induction therapy lacks prognostic significance, likely related to the delayed clearance of differentiating leukemic cells destined to eventually die.[74,75] However, the kinetics of molecular remission after completion of induction therapy is prognostic, with the persistence of minimal disease after three courses of therapy portending increased risk of relapse.[75,76,77]
Flow cytometric methods have been used for MRD detection and can detect leukemic blasts based on the expression of aberrant surface antigens that differ from the pattern observed in normal progenitors. In a COG analysis (AAML0531 [NCT00372593]) of 784 patients, 69% of patients (n = 544) were MRD negative (defined as <0.02%) in their bone marrow at the end of induction 1 (EOI1). Those patients had better disease-free survival rates (57%; 95% CI, 53%–61%; P < .001) and overall survival rates (73%; 95% CI, 69%–76%; P < .001) than patients who were MRD positive (DFS: 30%; 95% CI, 25%–36% and OS: 48%; 95% CI, 42%–54%).[67] Additionally, in the 76% of patients who were in morphological remission at EOI1, 20% were MRD positive and had a significantly worse outcome than the patients who were MRD negative/morphology negative. In the 24% of patients who were not in morphological remission, 36% were actually MRD negative and had significantly better outcomes than the patients who were MRD positive/morphology positive. This was also true in patients with marrow blast percentages in excess of 15%, 27% of whom had MRD-negative bone marrow and significantly better outcomes.[67] A CCG study of 252 pediatric patients with AML in morphological remission demonstrated that MRD as assessed by flow cytometry was the strongest prognostic factor predicting outcome in a multivariate analysis.[78] Other reports have confirmed both the utility of flow cytometric methods for MRD detection in the pediatric AML setting and the prognostic significance of MRD at various time points after treatment initiation.[62,63,65]
Risk Classification Systems
Risk classification for treatment assignment has been used by several cooperative groups performing clinical trials in children with AML. In the COG, stratifying therapeutic choices on the basis of risk factors is a relatively recent approach for the non-APL, non–Down syndrome patient. Classification is most directly derived from the observations of the MRC AML 10 trial for EFS and OS [61] and further applied based on the ability of the pediatric patient to undergo reinduction and obtain a second complete remission and their subsequent OS after first relapse.[79]
The following COG trials have used a risk classification system to stratify treatment choices:
- In COG AAML0531 (NCT00372593), the first COG trial to stratify therapy by risk group, patients were stratified into three risk groups on the basis of diagnostic cytogenetics and response after induction 1.[8]
- Low-risk patients included those diagnosed with a core-binding factor AML (either t(8;21) or inv(16)).
- High-risk patients had either monosomy 7, monosomy 5 or del5q, chromosome 3 abnormalities, or a poor response to induction 1 therapy with morphological marrow leukemic blasts (>15%).
- All other patients fell into the intermediate-risk category.
- This resulted in a risk distribution of 24% low risk, 59% intermediate risk, and 17% high risk.
- In the subsequent COG trial COG-AAML1031 (NCT01371981), the risk groups were reduced to two on the basis of the finding that those in the intermediate category could be more specifically and prognostically defined by adding the use of MRD by multiparameter flow cytometry.[22,80]
- Patients whose cytogenetics and/or molecular genetics were noninformative (i.e., traditional intermediate risk) and were negative for MRD (<0.1%) were placed in the low-risk category.
- Patients who were positive for MRD (≥0.1%) were placed in the high-risk category.
- In the COG-AAML1031 trial, the study stratification was further based on cytogenetics, molecular markers, and MRD at bone marrow recovery postinduction 1, with patients being divided into a low-risk or high-risk group as follows:[22]
- The low-risk group represented 78% of patients, had a 3-year OS rate from the end of induction 1 of 74.1% (±3.4%), and was defined by the following:
- Inv(16), t(8;21), NPM1 mutations, or CEBPA mutations regardless of MRD and other cytogenetics.
- Intermediate-risk cytogenetics (defined by the absence of either low-risk or high-risk cytogenetic characteristics) with negative MRD (<0.1% by flow cytometry) at end of induction 1.
- The high-risk group represented the remaining 22% of patients, had a 3-year OS rate from the end of induction 1 of 36.9% (±7.6%), and was defined by the following:
- High-allelic ratio FLT3 ITD positive with any MRD status.
- Monosomy 7 with any MRD status.
- Monosomy 5/del(5q) with any MRD status.
- Intermediate-risk cytogenetics with positive MRD at end of induction 1.
Where risk factors contradicted each other, the following evidence-based table was used (see Table 7).
Table 7. Risk Assignment in the AAML1031 Studya,b Risk Assignment: Low Risk High Risk Low-Risk Group 1 Low-Risk Group 2 High-Risk Group 1 High-Risk Group 2 High-Risk Group 3 ITD = internal tandem duplications. a Groups are based on combinations of risk factors, which may be found in any individual patient. bBold indicates the overriding risk factor in risk-group assignment. c NPM1,CEBPA, t(8;21), inv(16). d Monosomy 7, monosomy 5, del(5q). FLT3ITD allelic ratio Low/negative Low/negative High Low/negative Low/negative Good-risk molecular markersc Present Absent Any Absent Absent Poor-risk cytogenetic markersd Any Absent Any Present Absent Minimal residual disease Any Negative Any Any Positive
- The low-risk group represented 78% of patients, had a 3-year OS rate from the end of induction 1 of 74.1% (±3.4%), and was defined by the following:
The high-risk group of patients was guided to transplantation in first remission with the most appropriate available donor. Patients in the low-risk group were instructed to pursue transplantation if they relapsed.[63,81]
Risk factors used for stratification vary by pediatric and adult cooperative clinical trial groups and the prognostic impact of a given risk factor may vary in their significance depending on the backbone of therapy used. Other pediatric cooperative groups use some or all of these same factors, generally choosing risk factors that have been reproducible across numerous trials and sometimes including additional risk factors previously used in their risk group stratification approach.
References:
- Ebb DH, Weinstein HJ: Diagnosis and treatment of childhood acute myelogenous leukemia. Pediatr Clin North Am 44 (4): 847-62, 1997.
- Chan GC, Wang WC, Raimondi SC, et al.: Myelodysplastic syndrome in children: differentiation from acute myeloid leukemia with a low blast count. Leukemia 11 (2): 206-11, 1997.
- Cheson BD, Bennett JM, Kopecky KJ, et al.: Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 21 (24): 4642-9, 2003.
- Walter RB, Kantarjian HM, Huang X, et al.: Effect of complete remission and responses less than complete remission on survival in acute myeloid leukemia: a combined Eastern Cooperative Oncology Group, Southwest Oncology Group, and M. D. Anderson Cancer Center Study. J Clin Oncol 28 (10): 1766-71, 2010.
- Konopleva M, Cheng SC, Cortes JE, et al.: Independent prognostic significance of day 21 cytogenetic findings in newly-diagnosed acute myeloid leukemia or refractory anemia with excess blasts. Haematologica 88 (7): 733-6, 2003.
- Hann IM, Webb DK, Gibson BE, et al.: MRC trials in childhood acute myeloid leukaemia. Ann Hematol 83 (Suppl 1): S108-12, 2004.
- Gibson BE, Webb DK, Howman AJ, et al.: Results of a randomized trial in children with Acute Myeloid Leukaemia: medical research council AML12 trial. Br J Haematol 155 (3): 366-76, 2011.
- Gamis AS, Alonzo TA, Meshinchi S, et al.: Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children's Oncology Group trial AAML0531. J Clin Oncol 32 (27): 3021-32, 2014.
- Wells RJ, Woods WG, Buckley JD, et al.: Treatment of newly diagnosed children and adolescents with acute myeloid leukemia: a Childrens Cancer Group study. J Clin Oncol 12 (11): 2367-77, 1994.
- Perel Y, Auvrignon A, Leblanc T, et al.: Impact of addition of maintenance therapy to intensive induction and consolidation chemotherapy for childhood acute myeloblastic leukemia: results of a prospective randomized trial, LAME 89/91. Leucámie Aiqüe Myéloïde Enfant. J Clin Oncol 20 (12): 2774-82, 2002.
- Fenaux P, Chastang C, Chevret S, et al.: A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood 94 (4): 1192-200, 1999.
- Lo-Coco F, Avvisati G, Vignetti M, et al.: Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 369 (2): 111-21, 2013.
- Avvisati G, Lo-Coco F, Paoloni FP, et al.: AIDA 0493 protocol for newly diagnosed acute promyelocytic leukemia: very long-term results and role of maintenance. Blood 117 (18): 4716-25, 2011.
- Cooper TM, Franklin J, Gerbing RB, et al.: AAML03P1, a pilot study of the safety of gemtuzumab ozogamicin in combination with chemotherapy for newly diagnosed childhood acute myeloid leukemia: a report from the Children's Oncology Group. Cancer 118 (3): 761-9, 2012.
- Leung W, Hudson MM, Strickland DK, et al.: Late effects of treatment in survivors of childhood acute myeloid leukemia. J Clin Oncol 18 (18): 3273-9, 2000.
- Webb DK, Harrison G, Stevens RF, et al.: Relationships between age at diagnosis, clinical features, and outcome of therapy in children treated in the Medical Research Council AML 10 and 12 trials for acute myeloid leukemia. Blood 98 (6): 1714-20, 2001.
- Razzouk BI, Estey E, Pounds S, et al.: Impact of age on outcome of pediatric acute myeloid leukemia: a report from 2 institutions. Cancer 106 (11): 2495-502, 2006.
- Lange BJ, Smith FO, Feusner J, et al.: Outcomes in CCG-2961, a children's oncology group phase 3 trial for untreated pediatric acute myeloid leukemia: a report from the children's oncology group. Blood 111 (3): 1044-53, 2008.
- Creutzig U, Büchner T, Sauerland MC, et al.: Significance of age in acute myeloid leukemia patients younger than 30 years: a common analysis of the pediatric trials AML-BFM 93/98 and the adult trials AMLCG 92/99 and AMLSG HD93/98A. Cancer 112 (3): 562-71, 2008.
- Woods WG, Franklin AR, Alonzo TA, et al.: Outcome of adolescents and young adults with acute myeloid leukemia treated on COG trials compared to CALGB and SWOG trials. Cancer 119 (23): 4170-9, 2013.
- Canner J, Alonzo TA, Franklin J, et al.: Differences in outcomes of newly diagnosed acute myeloid leukemia for adolescent/young adult and younger patients: a report from the Children's Oncology Group. Cancer 119 (23): 4162-9, 2013.
- Aplenc R, Meshinchi S, Sung L, et al.: Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: a report from the Children's Oncology Group. Haematologica 105 (7): 1879-1886, 2020.
- Creutzig U, Zimmermann M, Bourquin JP, et al.: Favorable outcome in infants with AML after intensive first- and second-line treatment: an AML-BFM study group report. Leukemia 26 (4): 654-61, 2012.
- Kawasaki H, Isoyama K, Eguchi M, et al.: Superior outcome of infant acute myeloid leukemia with intensive chemotherapy: results of the Japan Infant Leukemia Study Group. Blood 98 (13): 3589-94, 2001.
- Masetti R, Rondelli R, Fagioli F, et al.: Infants with acute myeloid leukemia treated according to the Associazione Italiana di Ematologia e Oncologia Pediatrica 2002/01 protocol have an outcome comparable to that of older children. Haematologica 99 (8): e127-9, 2014.
- Guest EM, Aplenc R, Sung L, et al.: Gemtuzumab ozogamicin in infants with AML: results from the Children's Oncology Group trials AAML03P1 and AAML0531. Blood 130 (7): 943-945, 2017.
- Aplenc R, Alonzo TA, Gerbing RB, et al.: Ethnicity and survival in childhood acute myeloid leukemia: a report from the Children's Oncology Group. Blood 108 (1): 74-80, 2006.
- Conneely SE, McAtee CL, Gupta R, et al.: Association of race and ethnicity with clinical phenotype, genetics, and survival in pediatric acute myeloid leukemia. Blood Adv 5 (23): 4992-5001, 2021.
- Rubnitz JE, Lensing S, Razzouk BI, et al.: Effect of race on outcome of white and black children with acute myeloid leukemia: the St. Jude experience. Pediatr Blood Cancer 48 (1): 10-5, 2007.
- Lange BJ, Kobrinsky N, Barnard DR, et al.: Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: Children's Cancer Group Studies 2861 and 2891. Blood 91 (2): 608-15, 1998.
- Sorrell AD, Alonzo TA, Hilden JM, et al.: Favorable survival maintained in children who have myeloid leukemia associated with Down syndrome using reduced-dose chemotherapy on Children's Oncology Group trial A2971: a report from the Children's Oncology Group. Cancer 118 (19): 4806-14, 2012.
- Taub JW, Berman JN, Hitzler JK, et al.: Improved outcomes for myeloid leukemia of Down syndrome: a report from the Children's Oncology Group AAML0431 trial. Blood 129 (25): 3304-3313, 2017.
- Creutzig U, Reinhardt D, Diekamp S, et al.: AML patients with Down syndrome have a high cure rate with AML-BFM therapy with reduced dose intensity. Leukemia 19 (8): 1355-60, 2005.
- Massey GV, Zipursky A, Chang MN, et al.: A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood 107 (12): 4606-13, 2006.
- Gamis AS, Woods WG, Alonzo TA, et al.: Increased age at diagnosis has a significantly negative effect on outcome in children with Down syndrome and acute myeloid leukemia: a report from the Children's Cancer Group Study 2891. J Clin Oncol 21 (18): 3415-22, 2003.
- Uffmann M, Rasche M, Zimmermann M, et al.: Therapy reduction in patients with Down syndrome and myeloid leukemia: the international ML-DS 2006 trial. Blood 129 (25): 3314-3321, 2017.
- Lange BJ, Gerbing RB, Feusner J, et al.: Mortality in overweight and underweight children with acute myeloid leukemia. JAMA 293 (2): 203-11, 2005.
- Inaba H, Surprise HC, Pounds S, et al.: Effect of body mass index on the outcome of children with acute myeloid leukemia. Cancer 118 (23): 5989-96, 2012.
- Creutzig U, Zimmermann M, Ritter J, et al.: Definition of a standard-risk group in children with AML. Br J Haematol 104 (3): 630-9, 1999.
- Pession A, Masetti R, Rizzari C, et al.: Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia. Blood 122 (2): 170-8, 2013.
- Sung L, Aplenc R, Alonzo TA, et al.: Predictors and short-term outcomes of hyperleukocytosis in children with acute myeloid leukemia: a report from the Children's Oncology Group. Haematologica 97 (11): 1770-3, 2012.
- Testi AM, Biondi A, Lo Coco F, et al.: GIMEMA-AIEOPAIDA protocol for the treatment of newly diagnosed acute promyelocytic leukemia (APL) in children. Blood 106 (2): 447-53, 2005.
- Kutny MA, Alonzo TA, Gerbing RB, et al.: Arsenic Trioxide Consolidation Allows Anthracycline Dose Reduction for Pediatric Patients With Acute Promyelocytic Leukemia: Report From the Children's Oncology Group Phase III Historically Controlled Trial AAML0631. J Clin Oncol 35 (26): 3021-3029, 2017.
- de Botton S, Coiteux V, Chevret S, et al.: Outcome of childhood acute promyelocytic leukemia with all-trans-retinoic acid and chemotherapy. J Clin Oncol 22 (8): 1404-12, 2004.
- Ortega JJ, Madero L, Martín G, et al.: Treatment with all-trans retinoic acid and anthracycline monochemotherapy for children with acute promyelocytic leukemia: a multicenter study by the PETHEMA Group. J Clin Oncol 23 (30): 7632-40, 2005.
- Chisholm KM, Heerema-McKenney AE, Choi JK, et al.: Acute erythroid leukemia is enriched in NUP98 fusions: a report from the Children's Oncology Group. Blood Adv 4 (23): 6000-6008, 2020.
- Reinhardt D, Diekamp S, Langebrake C, et al.: Acute megakaryoblastic leukemia in children and adolescents, excluding Down's syndrome: improved outcome with intensified induction treatment. Leukemia 19 (8): 1495-6, 2005.
- Schweitzer J, Zimmermann M, Rasche M, et al.: Improved outcome of pediatric patients with acute megakaryoblastic leukemia in the AML-BFM 04 trial. Ann Hematol 94 (8): 1327-36, 2015.
- de Rooij JD, Masetti R, van den Heuvel-Eibrink MM, et al.: Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood 127 (26): 3424-30, 2016.
- de Rooij JD, Branstetter C, Ma J, et al.: Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet 49 (3): 451-456, 2017.
- Barbaric D, Alonzo TA, Gerbing RB, et al.: Minimally differentiated acute myeloid leukemia (FAB AML-M0) is associated with an adverse outcome in children: a report from the Children's Oncology Group, studies CCG-2891 and CCG-2961. Blood 109 (6): 2314-21, 2007.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Central nervous system disease in pediatric acute myeloid leukemia: A report from the Children's Oncology Group. Pediatr Blood Cancer 64 (12): , 2017.
- Abbott BL, Rubnitz JE, Tong X, et al.: Clinical significance of central nervous system involvement at diagnosis of pediatric acute myeloid leukemia: a single institution's experience. Leukemia 17 (11): 2090-6, 2003.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: The presence of central nervous system disease at diagnosis in pediatric acute myeloid leukemia does not affect survival: a Children's Oncology Group study. Pediatr Blood Cancer 55 (3): 414-20, 2010.
- Lugthart S, Gröschel S, Beverloo HB, et al.: Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol 28 (24): 3890-8, 2010.
- Creutzig U, van den Heuvel-Eibrink MM, Gibson B, et al.: Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 120 (16): 3187-205, 2012.
- Eidenschink Brodersen L, Alonzo TA, Menssen AJ, et al.: A recurrent immunophenotype at diagnosis independently identifies high-risk pediatric acute myeloid leukemia: a report from Children's Oncology Group. Leukemia 30 (10): 2077-2080, 2016.
- Pardo LM, Voigt AP, Alonzo TA, et al.: Deciphering the Significance of CD56 Expression in Pediatric Acute Myeloid Leukemia: A Report from the Children's Oncology Group. Cytometry B Clin Cytom 98 (1): 52-56, 2020.
- Smith JL, Ries RE, Hylkema T, et al.: Comprehensive Transcriptome Profiling of Cryptic CBFA2T3-GLIS2 Fusion-Positive AML Defines Novel Therapeutic Options: A COG and TARGET Pediatric AML Study. Clin Cancer Res 26 (3): 726-737, 2020.
- Lamble AJ, Eidenschink Brodersen L, Alonzo TA, et al.: CD123 Expression Is Associated With High-Risk Disease Characteristics in Childhood Acute Myeloid Leukemia: A Report From the Children's Oncology Group. J Clin Oncol 40 (3): 252-261, 2022.
- Wheatley K, Burnett AK, Goldstone AH, et al.: A simple, robust, validated and highly predictive index for the determination of risk-directed therapy in acute myeloid leukaemia derived from the MRC AML 10 trial. United Kingdom Medical Research Council's Adult and Childhood Leukaemia Working Parties. Br J Haematol 107 (1): 69-79, 1999.
- van der Velden VH, van der Sluijs-Geling A, Gibson BE, et al.: Clinical significance of flowcytometric minimal residual disease detection in pediatric acute myeloid leukemia patients treated according to the DCOG ANLL97/MRC AML12 protocol. Leukemia 24 (9): 1599-606, 2010.
- Loken MR, Alonzo TA, Pardo L, et al.: Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: a report from Children's Oncology Group. Blood 120 (8): 1581-8, 2012.
- Buldini B, Rizzati F, Masetti R, et al.: Prognostic significance of flow-cytometry evaluation of minimal residual disease in children with acute myeloid leukaemia treated according to the AIEOP-AML 2002/01 study protocol. Br J Haematol 177 (1): 116-126, 2017.
- Rubnitz JE, Inaba H, Dahl G, et al.: Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol 11 (6): 543-52, 2010.
- Tierens A, Bjørklund E, Siitonen S, et al.: Residual disease detected by flow cytometry is an independent predictor of survival in childhood acute myeloid leukaemia; results of the NOPHO-AML 2004 study. Br J Haematol 174 (4): 600-9, 2016.
- Brodersen LE, Gerbing RB, Pardo ML, et al.: Morphologic remission status is limited compared to ΔN flow cytometry: a Children's Oncology Group AAML0531 report. Blood Adv 4 (20): 5050-5061, 2020.
- Buonamici S, Ottaviani E, Testoni N, et al.: Real-time quantitation of minimal residual disease in inv(16)-positive acute myeloid leukemia may indicate risk for clinical relapse and may identify patients in a curable state. Blood 99 (2): 443-9, 2002.
- Viehmann S, Teigler-Schlegel A, Bruch J, et al.: Monitoring of minimal residual disease (MRD) by real-time quantitative reverse transcription PCR (RQ-RT-PCR) in childhood acute myeloid leukemia with AML1/ETO rearrangement. Leukemia 17 (6): 1130-6, 2003.
- Weisser M, Haferlach C, Hiddemann W, et al.: The quality of molecular response to chemotherapy is predictive for the outcome of AML1-ETO-positive AML and is independent of pretreatment risk factors. Leukemia 21 (6): 1177-82, 2007.
- Krönke J, Schlenk RF, Jensen KO, et al.: Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: a study from the German-Austrian acute myeloid leukemia study group. J Clin Oncol 29 (19): 2709-16, 2011.
- Corbacioglu A, Scholl C, Schlenk RF, et al.: Prognostic impact of minimal residual disease in CBFB-MYH11-positive acute myeloid leukemia. J Clin Oncol 28 (23): 3724-9, 2010.
- Cloos J, Goemans BF, Hess CJ, et al.: Stability and prognostic influence of FLT3 mutations in paired initial and relapsed AML samples. Leukemia 20 (7): 1217-20, 2006.
- Mandelli F, Diverio D, Avvisati G, et al.: Molecular remission in PML/RAR alpha-positive acute promyelocytic leukemia by combined all-trans retinoic acid and idarubicin (AIDA) therapy. Gruppo Italiano-Malattie Ematologiche Maligne dell'Adulto and Associazione Italiana di Ematologia ed Oncologia Pediatrica Cooperative Groups. Blood 90 (3): 1014-21, 1997.
- Burnett AK, Grimwade D, Solomon E, et al.: Presenting white blood cell count and kinetics of molecular remission predict prognosis in acute promyelocytic leukemia treated with all-trans retinoic acid: result of the Randomized MRC Trial. Blood 93 (12): 4131-43, 1999.
- Diverio D, Rossi V, Avvisati G, et al.: Early detection of relapse by prospective reverse transcriptase-polymerase chain reaction analysis of the PML/RARalpha fusion gene in patients with acute promyelocytic leukemia enrolled in the GIMEMA-AIEOP multicenter "AIDA" trial. GIMEMA-AIEOP Multicenter "AIDA" Trial. Blood 92 (3): 784-9, 1998.
- Martinelli G, Ottaviani E, Testoni N, et al.: Disappearance of PML/RAR alpha acute promyelocytic leukemia-associated transcript during consolidation chemotherapy. Haematologica 83 (11): 985-8, 1998.
- Sievers EL, Lange BJ, Alonzo TA, et al.: Immunophenotypic evidence of leukemia after induction therapy predicts relapse: results from a prospective Children's Cancer Group study of 252 patients with acute myeloid leukemia. Blood 101 (9): 3398-406, 2003.
- Webb DK, Wheatley K, Harrison G, et al.: Outcome for children with relapsed acute myeloid leukaemia following initial therapy in the Medical Research Council (MRC) AML 10 trial. MRC Childhood Leukaemia Working Party. Leukemia 13 (1): 25-31, 1999.
- Tarlock K, Meshinchi S: Pediatric acute myeloid leukemia: biology and therapeutic implications of genomic variants. Pediatr Clin North Am 62 (1): 75-93, 2015.
- Pui CH, Carroll WL, Meshinchi S, et al.: Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol 29 (5): 551-65, 2011.
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence has been slowly increasing since 1975.[1] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence.[2] This multidisciplinary team approach incorporates the skills of the following pediatric specialists and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life.
- Primary care physicians.
- Pediatric surgical subspecialists.
- Radiation oncologists.
- Pediatric medical oncologists/hematologists.
- Rehabilitation specialists.
- Pediatric nurse specialists.
- Social workers.
For specific information about supportive care for children and adolescents with cancer, see the summaries on Supportive and Palliative Care.
The American Academy of Pediatrics has outlined guidelines for pediatric cancer centers and their role in the treatment of children with cancer.[3] At these pediatric cancer centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate is offered to most patients and their families. Clinical trials for children and adolescents with cancer are generally designed to compare potentially better therapy with current standard therapy. Most of the progress made in identifying curative therapies for childhood cancers has been achieved through clinical trials. For more information about ongoing clinical trials, see the NCI website.
References:
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Wolfson J, Sun CL, Wyatt L, et al.: Adolescents and Young Adults with Acute Lymphoblastic Leukemia and Acute Myeloid Leukemia: Impact of Care at Specialized Cancer Centers on Survival Outcome. Cancer Epidemiol Biomarkers Prev 26 (3): 312-320, 2017.
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed May 19, 2023.
Treatment of Childhood AML
The general principles of therapy for children and adolescents with acute myeloid leukemia (AML) are discussed below, followed by a more specific discussion of the treatment of children with Down syndrome and acute promyelocytic leukemia (APL).
Overall survival (OS) rates have improved over the past three decades for children with AML, with 5-year survival rates now in the 55% to 65% range.[1,2,3,4] Overall remission-induction rates are approximately 85% to 90%, and event-free survival (EFS) rates from the time of diagnosis are in the 45% to 55% range.[2,3,4,5] There is, however, a wide range in outcome for different biological subtypes of AML; after taking specific biological factors of their leukemia into account, the predicted outcome for any individual patient may be much better or much worse than the overall outcome for the general population of children with AML. For more information, see the sections on Molecular Evaluation and Risk Classification Systems.
Induction Therapy
Contemporary pediatric AML protocols result in 85% to 90% complete remission (CR) rates.[6,7,8] Approximately 2% to 3% of patients die during the induction phase, most often caused by treatment-related complications.[6,7,8,9] To achieve a CR, inducing profound bone marrow aplasia (with the exception of the M3 APL subtype) is usually necessary with currently used combination-chemotherapy regimens. Because induction chemotherapy produces severe myelosuppression, morbidity and mortality from infection or hemorrhage during the induction period may be significant.
Treatment options for children with AML during the induction phase may include the following:
- Chemotherapy.
- Immunotherapeutic approaches (e.g., gemtuzumab ozogamicin).
- Targeted therapy (e.g., FLT3 inhibitors).
- Supportive care.
Chemotherapy
The two most effective and essential drugs used to induce remission in children with AML are cytarabine and an anthracycline. Commonly used pediatric induction therapy regimens use cytarabine and an anthracycline in combination with other agents such as etoposide and/or thioguanine.[3,10,11]
Evidence (induction chemotherapy regimen):
- The United Kingdom Medical Research Council (MRC) AML10 trial compared induction with cytarabine, daunorubicin, and etoposide (ADE) versus cytarabine and daunorubicin administered with thioguanine (DAT).[12]
- The results showed no difference in remission rate or disease-free survival (DFS) between the thioguanine and etoposide arms, although the thioguanine-containing regimen was associated with increased toxicity.
- The MRC AML15 trial demonstrated that induction with daunorubicin and cytarabine (DA) resulted in equivalent survival rates when compared with ADE induction.[13]
The anthracycline that has been most used in induction regimens for children with AML is daunorubicin,[3,10,11] although idarubicin and the anthracenedione mitoxantrone have also been used.[6,14,15] Randomized trials have attempted to determine whether any other anthracycline or anthracenedione is superior to daunorubicin as a component of induction therapy for children with AML. In the absence of convincing data that another anthracycline or mitoxantrone produces superior outcome over daunorubicin when given at an equitoxic dose, daunorubicin remains the anthracycline most commonly used during induction therapy for children with AML in the United States.
Evidence (anthracycline):
- The German Berlin-Frankfurt-Münster (BFM) Group AML-BFM 93 study evaluated cytarabine plus etoposide with either daunorubicin or idarubicin (ADE or AIE).[11,14]
- Similar EFS and OS rates were observed for both induction treatments.
- The MRC-LEUK-AML12 (NCT00002658) clinical trial studied induction with cytarabine, mitoxantrone, and etoposide (MAE) in children and adults with AML compared with a similar regimen using daunorubicin (ADE).[6,16]
- For all patients, the MAE regimen produced a reduction in relapse risk, but the increased rate of treatment-related mortality observed for patients receiving MAE led to no significant difference in DFS or OS rates when compared with ADE.[16]
- Similar results were noted when analyses were restricted to pediatric patients.[6]
- The AML-BFM 2004 (NCT00111345) clinical trial compared liposomal daunorubicin (L-DNR) with idarubicin at a higher-than-equivalent dose (80 mg/m2 vs. 12 mg/m2 per day for 3 days) during induction.[17]
- Five-year OS and EFS rates were similar in both treatment arms.
- Treatment-related mortality was significantly lower with L-DNR than with idarubicin (2 of 257 patients vs. 10 of 264 patients).
- The COG AAML1031 (NCT01371981) trial used mitoxantrone with high-dose cytarabine in its two-cycle induction phase for patients with high-risk AML.[18]
- In a planned comparison with the AAML0531 (NCT00372593) trial, which used a standard ADE regimen in the second induction cycle for similar patients, neither response nor survival was improved, whereas toxicity was increased in patients who received mitoxantrone.
Evidence (reduced-anthracycline induction regimen):
- Although the combination of an anthracycline and cytarabine is the basis of initial standard induction therapy for adults and children, there is evidence that alternative drugs can be used to reduce the use of anthracyclines when necessary. In the St. Jude Children's Research Hospital (SJCRH) AML08 (NCT00703820) protocol, patients were randomly assigned to receive either clofarabine/cytarabine (CA) or high-dose cytarabine combined with daunorubicin and etoposide (HD-ADE) for induction I; all patients then received the anthracycline-containing, standard-dose ADE regimen for induction II.[19]
- Despite a higher rate of minimal residual disease (MRD) in the CA group at day 22 of induction I (47% vs. 35%; P = .04), 3-year EFS and OS rates were similar between the two groups.
The intensity of induction therapy influences the overall outcome of therapy. The CCG-2891 study demonstrated that intensively timed induction therapy (4-day treatment courses separated by only 6 days) produced better EFS than standard-timing induction therapy (4-day treatment courses separated by 2 weeks or longer).[20] The MRC has intensified induction therapy by prolonging the duration of cytarabine treatment to 10 days.[10]
In adults, another method of intensifying induction therapy is to use high-dose cytarabine. While studies in nonelderly adults suggest an advantage for intensifying induction therapy with high-dose cytarabine (2–3 g/m2 /dose) compared with standard-dose cytarabine,[21] a benefit for the use of high-dose cytarabine in children was not observed using a cytarabine dose of 1 g/m2 given twice daily for 7 days with daunorubicin and thioguanine.[22] A second pediatric study also failed to detect a benefit for high-dose cytarabine over standard-dose cytarabine when used during induction therapy.[23]
Immunotherapeutic approaches
Because further intensification of induction regimens has increased toxicity with little improvement in EFS or OS, alternative approaches, such as the use of gemtuzumab ozogamicin, have been examined.
Antibody-drug conjugate therapy (gemtuzumab ozogamicin)
Gemtuzumab ozogamicin is a CD33-directed monoclonal antibody linked to a calicheamicin, a cytotoxic agent.
Evidence (gemtuzumab ozogamicin during induction):
- The Children's Oncology Group (COG) completed a series of trials—AAML03P1 (NCT00070174), a pilot study, and AAML0531 (NCT00372593), a randomized trial—that examined the incorporation of gemtuzumab ozogamicin into induction therapy.[8,9]
- With the use of gemtuzumab ozogamicin during induction cycle 1, dosed at 3 mg/m2 on day 6, the randomized trial identified an improvement in EFS but not in OS; this was because of a reduction in postremission relapse overall and specifically in distinct subsets of patients. These subsets included patients with low-risk cytogenetics, patients with intermediate-risk KMT2A-rearranged AML (with both high-risk and nonhigh-risk translocations) who went on to receive stem cell transplantation (SCT) from a matched-related donor,[24] and patients with high-risk high-allelic ratio (>0.4) FLT3 internal tandem duplication (ITD) AML who then received a SCT from any donor.[25]
- The efficacy and safety of gemtuzumab ozogamicin in children, which included infants as young as 1 month, was established in these trials.[26]
- Fractionated gemtuzumab ozogamicin dosing (3 mg/m2 per dose on days 1, 4, and 7; maximum dose, 5 mg), which has been shown to be safe and effective in adult patients with de novo AML, is an alternative option to single-dose administration during induction.[27] Because this is the recommended adult dosing method, this schedule is now being evaluated in the MyeChild 01 (NCT02724163) phase III study for pediatric patients with de novo AML in the United Kingdom.
- The characteristics of CD33, the target of gemtuzumab ozogamicin, have been examined to further identify the patients who will benefit most from this agent.
- The expression intensity of CD33 on leukemic cells appeared to predict which patients benefited from gemtuzumab ozogamicin on the COG AAML0531 clinical trial.[28][Level of evidence B1] Patients whose CD33 intensity fell into the highest three population quartiles benefited from treatment with gemtuzumab ozogamicin (i.e., improved relapse risk, DFS, and EFS), whereas those in the lowest quartile had no reduction in relapse risk, EFS, or OS. This impact was seen for low-, intermediate-, and high-risk patients.
- In a retrospective analysis of the ALFA-0701 (NCT00927498) trial of older adults, higher CD33 expression corresponded with greater benefit from treatment with gemtuzumab ozogamicin.[29]
- The CD33 receptor on AML cells exhibited architectural variability (polymorphism) that resulted in 51% of patients expressing the single nucleotide polymorphism (SNP) rs12459419 (designated CC), and these patients had a significant reduction in relapse with the use of gemtuzumab ozogamicin compared with patients who were not treated with this drug (26% vs. 49%; P < .001). The alteration of this SNP resulted in a CD33 isoform lacking the CD33 IgV domain to which gemtuzumab ozogamicin binds and that is used in diagnostic immunophenotyping.[30]
- For patients with either a one or two allele C>T mutation (CT and TT phenotypes, respectively) at this SNP, there was no reduction in relapse when adding gemtuzumab ozogamicin therapy (5-year cumulative incidence of relapse, 39% vs. 40%; P = .85).
- A meta-analysis of five randomized clinical trials that evaluated gemtuzumab ozogamicin for adults with AML observed the following:[31]
- The greatest OS benefit was for patients with low-risk cytogenetics (t(8;21)(q22;q22) and inv(16)(p13;q22)/t(16;16)(p13;q22)).
- Adult AML patients with intermediate-risk cytogenetics who received gemtuzumab ozogamicin had a significant but more modest improvement in OS.
- There was no evidence of benefit for patients with adverse cytogenetics.
- The evidence for a benefit in patients with FLT3 ITD mutations was mixed; the French ALFA-0701 (NCT00927498) trial showed a trend towards a benefit, whereas the five-trial meta-analysis study did not find a benefit.[27,31] These trials did not examine the outcomes specifically for the combination of gemtuzumab ozogamicin followed by SCT, as was reported by the COG.[25]
Targeted therapy
Similar to immunotherapeutic approaches, the use of targeted therapy attempts to circumvent the severe toxicity of traditional chemotherapy by employing agents that target leukemia-specific mutations and/or their abnormal present or missing byproducts. As opposed to adult AML (except in APL as described in a later section), randomized clinical trials have not yet demonstrated that targeted therapies improve outcomes in children with newly diagnosed AML; therefore, targeted therapies have not been incorporated into the standard therapeutic induction regimens outside of clinical trials. Because most data on the use of targeted agents are from adult clinical trials, the adult experience is initially described, followed by a description of the more limited experience in children.
FLT3 inhibitors in de novo AML
Because of the high prevalence of FLT3 mutations in adult AML and adverse impact in AML patients of all ages, the FLT3 target has received the greatest attention for target-specific drug development in AML. Among the various FLT3 inhibitors developed and clinically studied, midostaurin, a multikinase inhibitor, is the only one with U.S. Food and Drug Administration (FDA) approval for adult de novo AML; it was approved in 2017 for use with conventional backbone chemotherapy but not as a single agent.[32]
Midostaurin
Evidence (midostaurin for adults with de novo AML):
- In a randomized, placebo-controlled, phase III study (CALGB10603/RATIFY [NCT00651261]) of 717 adults aged 18 to 59 years with FLT3 ITD or TKD-mutated AML, standard chemotherapy was given with or without midostaurin (50 mg/dose twice daily) followed by maintenance midostaurin or placebo for patients who did not proceed to SCT.[33]
- OS (the primary endpoint) was significantly better for patients who received midostaurin (median OS, 74.7 months [95% confidence interval (CI), 31.5–not reached] vs. 25.6 months [95% CI, 18.6–42.9]; hazard ratio [HR], 0.78 [95% CI, 0.63–0.96]; P = .009), as was EFS (median EFS, 8.2 months [95% CI, 5.4–10.7] vs. 3.0 months [95% CI, 1.9–5.9]; HR, 0.78 [95% CI, 0.66–0.93]; P = .002).
- This benefit was seen across all FLT3 subgroups regardless of whether allogeneic SCT was utilized in consolidation.
- A second single-arm trial in 284 adults (aged 18–70 years) with FLT3 ITD AML added midostaurin (50 mg/dose twice daily) to intensive chemotherapy followed by allogeneic SCT or consolidation, and all patients had a subsequent midostaurin maintenance phase.[34]
- The 2-year EFS rate was 37.7% (95% CI, 32%–44.3%), and the OS rate was 50.9% (95% CI, 44.9%–57.6%).
- Using a historical-control comparison, significant improvement in EFS was reported (HR, 0.58; 95% CI, 0.48–0.70; P < .001).
Midostaurin has been studied in children with relapsed/refractory AML,[35] but there is no experience with midostaurin in children with newly diagnosed AML. For more information, see the Targeted therapy (FLT3 inhibitors) section.
Sorafenib
Sorafenib, another multikinase inhibitor, has been approved for the treatment of other malignancies, but it has not been approved for use in AML. This agent has been evaluated for use in adult and pediatric patients with de novo FLT3-mutated AML.
Evidence (sorafenib):
- Sorafenib was shown to improve EFS in the COG AAML1031 (NCT01371981) study of pediatric patients with de novo AML and high-allelic ratio (HAR) (i.e., >0.4) FLT3 ITD mutations. Seventy-two patients who received sorafenib were evaluable for response. The patients in this study were compared with patients with AML and HAR FLT3 ITD (N = 76) in the AAML1031 and COG AAML0531 trials who did not receive sorafenib.[36]
- The morphological CR rate after induction cycle I significantly improved for patients who received sorafenib (75% vs. 57%; P = .028).
- However, there was a similar prevalence of residual MRD in both groups of patients (48% vs. 45%; P = .724).
- Patients who received sorafenib had significantly improved 3-year EFS rates (55.9% vs. 31.9%; P = .001), DFS rates (70.9% vs. 49.4%; P = .032), and relapses after CR (17.6% vs. 44.1%; P = .012).
- The OS rate did not improve after treatment with sorafenib (65.8% vs. 55.3%; P = .244).
- Although similar trends were seen in patients with AML harboring both HAR FLT3 ITD mutations and NPM1 mutations, they did not approach a significant level of benefit.
- Statistics showed that a benefit of sorafenib treatment remained after multivariable analyses that controlled for both NPM1 status and hematopoietic stem cell transplantation (HSCT) as a time-varying covariate.
Supportive care
In children with AML receiving modern intensive therapy, the estimated incidence of severe bacterial infections is 50% to 60%, and the estimated incidence of invasive fungal infections is 7.0% to 12.5%.[37,38,39] Several approaches have been examined to reduce the morbidity and mortality from infection in children with AML.
Hematopoietic growth factors
Hematopoietic growth factors such as granulocyte-macrophage colony-stimulating factor (GM-CSF) or granulocyte colony-stimulating factor (G-CSF) during AML induction therapy have been evaluated in multiple placebo-controlled studies in adults with AML in attempts to reduce the toxicity associated with prolonged myelosuppression.[7] These studies have generally shown a reduction in the duration of neutropenia of several days with the use of either G-CSF or GM-CSF [40] but have not shown significant effects on treatment-related mortality or OS.[40] For more information, see the Treatment Option Overview for AML section in Acute Myeloid Leukemia Treatment.
Routine prophylactic use of hematopoietic growth factors is not recommended for children with AML.
Evidence (against the use of hematopoietic growth factors):
- A randomized study in children with AML that evaluated G-CSF administered after induction chemotherapy showed a reduction in duration of neutropenia but no difference in infectious complications or mortality.[41]
- A higher relapse rate has been reported for children with AML expressing the differentiation defective G-CSF receptor isoform IV.[42]
Antimicrobial prophylaxis
The use of antibacterial prophylaxis in children undergoing treatment for AML has been supported by several studies. Studies, including one prospective randomized trial, suggest a benefit to the use of antibiotic prophylaxis.
Evidence (antimicrobial prophylaxis):
- A retrospective study from SJCRH in patients with AML reported that the use of intravenous (IV) cefepime or vancomycin in conjunction with oral ciprofloxacin or a cephalosporin significantly reduced the incidence of bacterial infection and sepsis compared with patients receiving only oral or no antibiotic prophylaxis.[43]
- The SJCRH results were confirmed in a subsequent study.[44]
- A retrospective report from the COG AAML0531 (NCT00372593) trial demonstrated significant reductions in sterile-site bacterial infection and particularly gram-positive, sterile-site infections with the use of antibacterial prophylaxis.[45] This study also reported that prophylactic use of G-CSF reduced bacterial and Clostridium difficile infections.[45]
- In a study that compared the percentage of bloodstream infections or invasive fungal infections in children with acute lymphoblastic leukemia (ALL) or AML who underwent chemotherapy and received antibacterial and antifungal prophylaxis, a significant reduction in both variables was observed when compared with a historical control group that did not receive any prophylaxis.[46]
- In the prospective COG ACCL0934 trial for children receiving intensive chemotherapy, patients were enrolled in two separate groups—patients with acute leukemia (consisting of AML or relapsed ALL) and patients undergoing SCT. Patients with acute leukemia were randomly assigned to receive levofloxacin (n = 96) or no prophylactic antibiotic (n = 99) during the period of neutropenia in one to two cycles of chemotherapy.[47]
- Analysis of the 195 children with acute leukemia revealed a significant reduction in bacteremia (43.4% to 21.9%, P = .001) and neutropenic fever (82.1% to 71.2%, P = .002) in the levofloxacin prophylaxis group compared with the control group, without increases in fungal infections, C. difficile–associated diarrhea, or musculoskeletal toxicities.
- There was no significant decrease in severe infections (3.6% vs. 5.9%, P = .20), and no bacterial infection–related deaths occurred in either group.
- Levofloxacin prophylaxis is consistent with the guidelines published by the American Society of Clinical Oncology and Infectious Diseases Society of America in 2018 for adult cancer patients considered at high risk of infection by virtue of neutropenia (<100 neutrophils/µL) in excess of 7 days.[48]
Antifungal prophylaxis
Antifungal prophylaxis is important in the management of patients with AML.
Evidence (antifungal prophylaxis):
- Two meta-analysis reports have suggested that antifungal prophylaxis in pediatric patients with AML during treatment-induced neutropenia or during bone marrow transplantation reduces the frequency of invasive fungal infections and, in some instances, nonrelapse mortality.[49,50]
- Another study surveyed institutions that enrolled patients on the COG AAML0531 (NCT00372593) trial and investigated if these institutions routinely prescribed antifungal prophylaxis.[45]
- The study found that antifungal prophylaxis did not reduce fungal infections or nonrelapse mortality.
- The study was limited, however, because the investigators did not analyze whether individual patients received antifungal prophylaxis, regardless of institutional guidance.
- Several randomized trials in adults with AML have reported significant benefit in reducing invasive fungal infection with the use of antifungal prophylaxis. Such studies have also balanced cost with adverse side effects; when effectiveness at reducing invasive fungal infection is balanced with these other factors, posaconazole, voriconazole, caspofungin, and micafungin are considered reasonable choices.[46,51,52,53,54,55]
- There is a single randomized study comparing two antifungal agents for prophylaxis in pediatric patients with AML. The COG ACCL0933 (NCT01307579) trial randomly assigned patients to receive prophylactic treatment with either fluconazole or caspofungin (an echinocandin with broader antiyeast and antimold activity than fluconazole).[56]
- Caspofungin was superior to fluconazole in achieving lower 5-month cumulative incidences of both proven or probable invasive fungal disease (3.1% vs. 7.2%; P = .03) and proven or probable invasive aspergillosis (0.5% vs. 3.1%; P = .046).
Cardiac monitoring
Bacteremia or sepsis and anthracycline use have been identified as significant risk factors in the development of cardiotoxicity, manifested as reduced left ventricular function.[57,58] Monitoring of cardiac function through the use of serial exams during therapy is an effective method for detecting cardiotoxicity and adjusting therapy as indicated. The use of dexrazoxane in conjunction with bolus dosing of anthracyclines can be an effective method of reducing the risk of cardiac dysfunction during therapy.[59]
Evidence (cardiac monitoring/dexrazoxane impact):
- In the COG AAML0531 (NCT00372593) trial, 8.6% of enrolled patients experienced left ventricular systolic dysfunction (LVSD) during protocol therapy, with a cumulative incidence of LVSD of 12% within 5 years of completing therapy.[57]
- Risk factors for LVSD during therapy included Black race, older age, underweight body mass, and bacteremia.
- The occurrence of LVSD adversely impacted 5-year EFS (HR, 1.57; 95% CI, 1.16–2.14; P = .004) and OS (HR, 1.59; 95% CI, 1.15–2.19; P = .005), which was primarily a result of nonrelapse mortality.
- In patients who experienced LVSD during therapy, there was a 12-fold greater risk of LVSD in the 5 years after the completion of therapy.
- The use of dexrazoxane was assessed in patients enrolled on the COG AAML1031 (NCT01371981) trial.[59]
- This trial mandated prospective cardiac monitoring with each cycle and in follow-up and found a higher LVSD incidence (39%) occurring at a median of 3.8 months from enrollment (interquartile range, 2–6.2 months) than was seen in the preceding trial.
- Among the approximately 10% of children (96 of 1,014) who electively received dexrazoxane with each dose of anthracycline, the incidence of LVSD (defined as ejection fraction <55% or shortening fraction <28%) was significantly less in these patients (26.5% vs. 42.2%; HR, 0.55; 95% CI, 0.36–0.86; P = .009) than in the patients who did not elect to receive dexrazoxane. This was also evident for risk of LVSD grade 2 or higher (60% lower). Patients who received dexrazoxane also had persistently better cardiac function after therapy (median follow-up, 3.5 years).
- Patients who received dexrazoxane had a lower treatment-related mortality (5.7% vs. 12.7%; P = .068), although the improved OS, EFS, and RR outcomes did not reach statistical significance.
Hospitalization
Hospitalization until adequate granulocyte (absolute neutrophil or phagocyte count) recovery has been used to reduce treatment-related mortality. The COG-2961 (NCT00002798) trial was the first to note a significant reduction in treatment-related mortality (19% before mandatory hospitalization was instituted in the trial along with other supportive care changes vs. 12% afterward); OS was also improved in this trial (P <.001).[3] Another analysis of the impact of hospitalization using a survey of institutional routine practice found that those who mandated hospitalization had nonsignificant reduction in patients' treatment-related mortality (adjusted HR, 0.60 [0.26–1.36, P = .22]) compared with institutions who had no set policy.[45] Although there was no significant benefit seen in this study, the authors noted the limitations, including its methodology (survey), an inability to validate cases, and limited power to detect differences in treatment-related mortality. To avoid prolonged hospitalizations until count recovery, some institutions have used outpatient IV antibiotic prophylaxis effectively.[44]
Induction failure (refractory AML)
Induction failure (the morphological presence of 5% or greater marrow blasts at the end of all induction courses) is seen in 10% to 15% of children with AML. Subsequent outcomes for patients with induction failure are similar to those for patients with AML who relapse early (<12 months after remission).[60,61]
Granulocytic sarcoma/chloroma
Granulocytic sarcoma (chloroma) describes extramedullary collections of leukemia cells. These collections can occur, albeit rarely, as the sole evidence of leukemia. In a review of three AML studies conducted by the former Children's Cancer Group, fewer than 1% of patients had isolated granulocytic sarcoma, and 11% had granulocytic sarcoma along with marrow disease at the time of diagnosis.[62] This incidence was also seen in the NOPHO-AML 2004 (NCT00476541) trial.[63]
Importantly, the patient who presents with an isolated tumor, without evidence of marrow involvement, must be treated as if there is systemic disease. Patients with isolated granulocytic sarcoma have a good prognosis if treated with current AML therapy.[62]
In a study of 1,459 children with newly diagnosed AML, patients with orbital granulocytic sarcoma and central nervous system (CNS) granulocytic sarcoma had better survival than did patients with marrow disease and granulocytic sarcoma at other sites and AML patients without any extramedullary disease.[63,64] Most patients with orbital granulocytic sarcoma have a t(8;21) abnormality, which has been associated with a favorable prognosis. The use of radiation therapy does not improve survival in patients with granulocytic sarcoma who have a complete response to chemotherapy, but may be necessary if the site(s) of granulocytic sarcoma do not show complete response to chemotherapy or for disease that recurs locally.[62]
Central Nervous System (CNS) Prophylaxis for AML
CNS involvement in patients with AML and its impact on prognosis has been discussed above in the Prognostic Factors in Childhood AML section. Therapy with either radiation or intrathecal chemotherapy has been used to treat CNS leukemia present at diagnosis and to prevent later development of CNS leukemia. The use of radiation has essentially been abandoned as a means of prophylaxis because of the lack of documented benefit and long-term sequelae.[65] The COG has used single-agent cytarabine for both CNS prophylaxis and therapy. Other groups have attempted to prevent CNS relapse by using additional intrathecal agents.
Evidence (CNS prophylaxis):
- The COG AAML0531 (NCT00372593) trial used single-agent cytarabine for prophylaxis.[66]
- As opposed to the low relapse rate associated with CNS1 disease (3.9%) seen in the 71% of enrolled patients, patients who had minimal evidence of CNS leukemia at diagnosis (CNS2 or blasts present when CSF WBC was <5 cells/HPF; 16% of newly diagnosed patients) were given twice-weekly intrathecal cytarabine until the CSF cleared. For the 96% of CNS2 patients whose CSF (95.8%) was cleared of leukemic blasts, 11.7% later experienced CNS relapse.
- CNS3 involvement at diagnosis (13%) conferred even worse outcomes. Despite clearing of leukemic blasts in 90.7% of children, 17.7% later experienced a CNS relapse. In a multivariate analysis, the presence of CNS3 involvement significantly worsened isolated CNS relapse risk (HR, 7.82; P = .003).
- Another methodology uses additional intrathecal agents, including triples, a combination of intrathecal cytarabine, hydrocortisone, and methotrexate.[67]
- The SJCRH reported that after switching from triples (their previous standard treatment) to single-agent cytarabine, the incidence of isolated CNS relapse increased from 0% (0 of 131 patients) to 9% (3 of 33 patients), prompting them to return to triples, which then reproduced a 0% (0 of 79 patients) CNS relapse rate.
Postremission Therapy for AML
A major challenge in the treatment of children with AML is to prolong the duration of the initial remission with additional chemotherapy or HSCT.
Treatment options for children with AML in postremission may include the following:
- Chemotherapy.
- HSCT.
- Targeted therapy (e.g., FLT3 inhibitors).[68]
Chemotherapy
Postremission chemotherapy includes some of the drugs used in induction while also introducing non–cross-resistant drugs and, commonly, high-dose cytarabine. Studies in adults with AML have demonstrated that consolidation with a high-dose cytarabine regimen improves outcome compared with consolidation with a standard-dose cytarabine regimen, particularly in patients with inv(16) and t(8;21) AML subtypes.[69] For more information, see the Treatment of AML in Remission section in Acute Myeloid Leukemia Treatment. Randomized studies evaluating the contribution of high-dose cytarabine to postremission therapy have not been conducted in children, but studies employing historical controls suggest that consolidation with a high-dose cytarabine regimen improves outcome compared with less-intensive consolidation therapies.[11,70,71]
The optimal number of postremission courses of therapy remains unclear, but appears to require at least two to three courses of intensive therapy after induction.[3]
Evidence (number of postremission courses of chemotherapy):
- A United Kingdom Medical Research Council (MRC) study randomly assigned adult and pediatric patients to either four or five courses of intensive therapy.[6,16][Level of evidence A1]
- Five courses of therapy did not show an advantage for relapse-free survival and OS.
- Based on this MRC data, in the COG AAML1031 (NCT01371981) trial, non–high-risk patients treated without HSCT in first CR (73% of all patients) received four cycles of chemotherapy (two induction cycles and two consolidation cycles) rather than five cycles (two induction cycles and three consolidation cycles); in the previous COG AAML0531 (NCT00372593) and AAML03P1 (NCT00070174) trials, patients who did not undergo HSCT received five cycles of chemotherapy.[72]
- In a retrospective analysis, non–high-risk patients treated without HSCT on the COG AAML1031 trial (four chemotherapy cycles) had significantly worse outcomes than did those who had received five cycles of chemotherapy on the AAML0531 trial (four- vs. five-cycle outcomes):
- The OS rate was 77.0% for patients who received four chemotherapy cycles, compared with 83.5% for patients who received five chemotherapy cycles (HR, 1.45; 95% CI, 0.97–2.17; P = .068).
- The DFS rate was 56% for patients who received four cycles, compared with 67% for patients who received five cycles (HR, 1.45; 95% CI, 1.10–1.91; P = .009).
- The relapse rate was 40.9% for patients who received four cycles, compared with 31.4% for patients who received five cycles (HR, 1.40; 95% CI, 1.06–1.85; P = .019).
- An exception was found in the low-risk subgroup defined by favorable cytogenetics or molecular genetics who were MRD negative at the end of induction cycle 1. This subset of patients had similar outcomes regardless of whether they received four chemotherapy cycles (AAML1031) or five chemotherapy cycles (AAML0531).
Additional study of the number of intensification courses and specific agents used will better address this issue, but these data suggest that four chemotherapy courses should only be administered to the favorable group described above, and that all other patients who do not undergo HSCT should receive five chemotherapy courses.
- In a retrospective analysis, non–high-risk patients treated without HSCT on the COG AAML1031 trial (four chemotherapy cycles) had significantly worse outcomes than did those who had received five cycles of chemotherapy on the AAML0531 trial (four- vs. five-cycle outcomes):
HSCT
The use of HSCT in first remission has been under evaluation since the late 1970s, and evidence-based appraisals concerning indications for autologous and allogeneic HSCT have been published. Prospective trials of transplantation in children with AML suggest that overall, 60% to 70% of children with HLA-matched donors available who undergo allogeneic HSCT during their first remission experience long-term remissions,[10,73] with the caveat that outcome after allogeneic HSCT is dependent on risk-classification status.[74]
In prospective trials that compared allogeneic HSCT with chemotherapy and/or autologous HSCT, superior DFS rates were observed for patients who were assigned to allogeneic HSCT on the basis of family 6/6 or 5/6 HLA-matched donors in adults and children.[10,73,75,76,77,78,79] However, the superiority of allogeneic HSCT over chemotherapy has not always been observed.[80] Several large cooperative group clinical trials for children with AML have found no benefit for autologous HSCT over intensive chemotherapy.[10,73,75,77]
Current application of allogeneic HSCT involves incorporation of risk classification to determine whether transplantation should be pursued in first remission. Because of the improved outcome in patients with favorable prognostic features (low-risk cytogenetic or molecular mutations) receiving contemporary chemotherapy regimens and the lack of demonstrable superiority for HSCT in this patient population, this group of patients typically receives matched-family donor (MFD) HSCT only after first relapse and the achievement of a second CR.[74,81,82,83]
An analysis from the Center for International Blood and Marrow Transplant Research (CIBMTR) examined pretransplant variables to create a model for predicting leukemia-free survival (LFS) posttransplant in pediatric patients (aged <18 years). All patients were first transplant recipients who had myeloablative conditioning, and all stem cells sources were included. For patients with AML, the predictors associated with lower LFS included age younger than 3 years, intermediate-risk or poor-risk cytogenetics, and second CR or higher with MRD positivity or not in CR. A scale was established to stratify patients on the basis of risk factors to predict survival. The 5-year LFS rate was 78% for the low-risk group, 53% for the intermediate-risk group, 40% for the high-risk group, and 25% for the very high-risk group.[84]
There is conflicting evidence regarding the role of allogeneic HSCT in first remission for patients with intermediate-risk characteristics (neither low-risk or high-risk cytogenetics or molecular mutations):
Evidence (allogeneic HSCT in first remission for patients with intermediate-risk AML):
- A study combining the results of the POG-8821, CCG-2891, COG-2961 (NCT00002798), and MRC AML10 studies identified a DFS and OS advantage for allogeneic HSCT in patients with intermediate-risk AML but not favorable-risk (inv(16) and t(8;21)) or poor-risk AML (del(5q), monosomy 5 or 7, or more than 15% blasts after first induction for POG/CCG studies); the MRC study included patients with 3q abnormalities and complex cytogenetics in the high-risk category.[74] Weaknesses of this study include the large percentage of patients not assigned to a risk group and the relatively low EFS and OS rates for patients with intermediate-risk AML assigned to chemotherapy, as compared with results observed in more recent clinical trials.[6,17]
- The AML99 clinical trial from the Japanese Childhood AML Cooperative Study Group observed a significant difference in DFS for intermediate-risk patients assigned to MFD HSCT, but there was not a significant difference in OS.[85]
- The AML-BFM 99 clinical trial demonstrated no significant difference in either DFS or OS for intermediate-risk patients assigned to MFD HSCT versus those assigned to chemotherapy.[80]
Given the improved outcome for patients with intermediate-risk AML in recent clinical trials and the burden of acute and chronic toxicities associated with allogeneic transplantation, many childhood AML treatment groups (including the COG) employ chemotherapy for intermediate-risk patients in first remission and reserve allogeneic HSCT for use after potential relapse.[6,85,86]
There are conflicting data regarding the role of allogeneic HSCT in first remission for patients with high-risk disease, complicated by the differing definitions of high risk used by different study groups.
Evidence (allogeneic HSCT in first remission for patients with high-risk AML):
- A retrospective analysis from the COG and CIBMTR compared chemotherapy only with matched-related donor and matched-unrelated donor HSCT for patients with AML and high-risk cytogenetics, defined as monosomy 7/del(7q), monosomy 5/del(5q), abnormalities of 3q, t(6;9), or complex karyotypes.[87]
- The analysis demonstrated no difference in the 5-year OS among the three treatment groups.
- A Nordic Society for Pediatric Hematology and Oncology study reported that time-intensive reinduction therapy followed by transplant with best available donor for patients whose AML did not respond to induction therapy resulted in 70% survival at a median follow-up of 2.6 years.[88][Level of evidence B4]
- A single-institution retrospective study of 36 consecutive patients (aged 0–30 years) with high-risk AML (FLT3 ITD, 11q23 KMT2A rearrangements, presence of chromosome 5 or 7 abnormalities, induction failure, persistent disease), who were in a morphological first remission before allogeneic transplant.[89]
- The investigators reported a 5-year OS rate of 72% and a LFS rate (from the time of transplant) of 69% with the use of a myeloablative conditioning regimen.
- They also reported a treatment-related mortality rate of 17%.
- These outcomes were similar to 14 standard-risk AML patients who underwent transplantation during the same time period.
- A subgroup analysis from the AML-BFM 98 clinical trial demonstrated improved survival rates for patients with 11q23 aberrations allocated to allogeneic HSCT, but not for patients without 11q23 aberrations.[80]
- For children with FLT3 ITD (high-allelic ratio), patients who received MFD HSCT (n = 6) had higher OS rates than did patients who received standard chemotherapy (n = 28); however, the number of cases studied limited the ability to draw conclusions.[90]
- A subsequent retrospective report from three consecutive trials in young adults with AML found that patients with FLT3 ITD high-allelic ratio benefited from allogeneic HSCT (P =.03), whereas patients with low-allelic ratio did not (P = .64).[91]
- A subset analysis of the COG phase III trial evaluated gemtuzumab ozogamicin during induction therapy in children with newly diagnosed AML.[25]
- For patients with FLT3 ITD high-allelic ratio who received HSCT, a lower relapse rate was observed for those who also received gemtuzumab ozogamicin (15% vs. 53%, P = .007).
- Conversely, patients who received gemtuzumab ozogamicin had higher rates of treatment-related mortality (19% vs. 7%, P = .08), resulting in overall improved DFS (65% vs. 40%, P = .08).
Many, but not all, pediatric clinical trial groups prescribe allogeneic HSCT for high-risk patients in first remission.[83] For example, the COG frontline AML clinical trial (COG-AAML1031) prescribes allogeneic HSCT in first remission only for patients with predicted high risk of treatment failure based on unfavorable cytogenetic and molecular characteristics and elevated end-of-induction MRD levels. On the other hand, the AML-BFM trials restrict allogeneic HSCT to patients in second CR and to refractory AML. This was based on results from their AML-BFM 98 study, which found no improvement in DFS or OS for high-risk patients receiving allogeneic HSCT in first CR, as well as the successful treatment using HSCT for a substantial proportion of patients who achieved a second CR.[80,92] Additionally, late sequelae (e.g., cardiomyopathy, skeletal anomalies, and liver dysfunction or cirrhosis) were increased for children undergoing allogeneic HSCT in first remission on the AML-BFM 98 study.[80]
Because definitions of high-, intermediate-, and low-risk AML are evolving because of the ongoing association of molecular characteristics of the tumor with outcome (e.g., FLT3 ITD, WT1 mutations, and NPM1 mutations) and response to therapy (e.g., MRD assessments postinduction therapy), further analysis of subpopulations of patients treated with allogeneic HSCT will be an ongoing need in current and future clinical trials.
If transplant is chosen in first CR, the optimal preparative regimen and source of donor cells has not been determined, although alternative donor sources, including haploidentical donors, are being studied.[79,93,94] There are no data that suggest total-body irradiation (TBI) is superior to busulfan-based myeloablative regimens.[80,81] Additionally, outstanding outcomes have been noted for patients who were treated with treosulfan-based regimens; however, trials comparing treosulfan with busulfan or TBI are lacking.[95]
Evidence (myeloablative regimen):
- A randomized trial that compared busulfan plus fludarabine with busulfan plus cyclophosphamide as a preparative regimen for AML in first CR demonstrated that the former regimen was associated with less toxicity and comparable DFS and OS.[96]
- In addition, a large prospective CIBMTR cohort study of children and adults with AML, myelodysplastic syndromes (MDS), and chronic myelogenous leukemia (CML) showed superior survival of patients with early-stage disease (chronic-phase CML, first CR AML, and MDS-refractory anemia) with busulfan-based regimens compared with TBI.[97]
- A CIBMTR study of 624 children with de novo AML who underwent transplantation between 2008 and 2016 and received either a TBI-based regimen (n = 199) or non-TBI–containing regimen (n = 425) demonstrated the following results:[98]
- TBI recipients had a higher nonrelapse mortality (P < .0001) with lower relapse (P < .0001), culminating in equivalent LFS and OS rates.
- TBI recipients experienced more grades 2 to 3 acute GVHD (56% vs. 27%; P < .0001) but had equivalent chronic GVHD incidence.
- TBI recipient survivors had a greater incidence of gonadal or growth deficiency (24% vs. 8%; P < .0001), but there were no differences seen in pulmonary, cardiac, or renal impairment.
Other than the APL subtype, there are no data that demonstrate that maintenance therapy given after intensive postremission therapy significantly prolongs remission duration. Maintenance chemotherapy failed to show benefit in two randomized studies that used modern intensive consolidation therapy,[70,99] and maintenance therapy with interleukin-2 also proved ineffective.[3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Recurrent or Refractory Childhood AML and Other Myeloid Malignancies
The diagnosis of recurrent or relapsed AML according to COG criteria is essentially the same as the criteria for making the diagnosis of AML. Usually this is defined as patients having more than 5% bone marrow blasts who were in previous remission after therapy for a diagnosis of AML according to WHO classification criteria.[100,101]
Despite second remission induction in over one-half of children with AML treated with drugs similar to drugs used in initial induction therapy, the prognosis for a child with recurrent or progressive AML is generally poor.[60,102]
Recurrent childhood AML
Approximately 50% to 60% of relapses occur within the first year after diagnosis, with most relapses occurring by 4 years from diagnosis.[102] The vast majority of relapses occur in the bone marrow, and CNS relapse is very uncommon.[102]
Prognosis and prognostic factors
Factors affecting the ability to attain a second remission include the following:
- Length of first remission. Length of first remission is an important factor affecting the ability to attain a second remission; children with a first remission of less than 1 year have substantially lower rates of second remission (50%–60%) than children whose first remission is greater than 1 year (70%–90%).[60,103,104] Survival for children with shorter first remissions is also substantially lower (approximately 10%) than that for children with first remissions exceeding 1 year (approximately 40%).[60,103,104,105] The Therapeutic Advances in Childhood Leukemia and Lymphoma Consortium also identified duration of previous remission as a powerful prognostic factor, with 5-year OS rates of 54% (± 10%) for patients with greater than 12 months first remission duration and 19% (± 6%) for patients with shorter periods of first remission.[106] In this same analysis, outcomes, primarily in early relapsing patients, declined with each attempt to reinduce remission (56% ± 5%, 25% ± 8%, and 17% ± 7% for each consecutive attempt).
- Molecular alterations. In addition, specific molecular alterations at the time of relapse have been reported to impact subsequent survival. For instance, the presence of either WT1 or FLT3 ITD mutations at first relapse were associated, as independent risk factors, with worse OS in patients achieving a second remission.[107]
Additional prognostic factors were identified in the following studies:
- In a report of 379 children with AML who relapsed after initial treatment on the German BFM group protocols, a second complete remission rate was 63% and the OS rate was 23%.[108][Level of evidence C1] The most significant prognostic factors associated with a favorable outcome after relapse included achieving second complete remission, a relapse greater than 12 months from initial diagnosis, no allogeneic bone marrow transplant in first remission, and favorable cytogenetics (t(8;21), t(15;17), and inv(16)).
- The international Relapsed AML 2001/01 (NCT00186966) trial also found that early response to salvage therapy was highly prognostic.[109][Level of evidence C2]
- A retrospective study of 71 patients with relapsed AML from Japan reported a 5-year OS rate of 37%. Patients who had an early relapse had a 27% second remission rate compared with 88% for patients who had a late relapse. The 5-year OS rate was higher in patients who underwent HSCT after achieving a second complete remission (66%) than in patients not in remission (17%).[105]
- Patients who relapsed on two consecutive Nordic Society of Pediatric Hematology and Oncology (NOPHO) AML trials between 1993 to 2012 were analyzed for survival (208 patients relapsed of 543 children initially treated). Second remissions were achieved in 146 children (70%) with a variety of reinduction regimens. The 5-year OS rate was 39%. Favorable prognostic factors included late relapse (≥1 year from diagnosis), no HSCT in first remission, and a core-binding factor AML subtype. For the children in second remission who underwent HSCT, the 5-year OS rate was 61%, as opposed to a 5-year OS rate of 18% for those who did not include HSCT in their therapy (P < .001).[110]
Treatment of recurrent AML
Treatment options for children with recurrent AML may include the following:
- Chemotherapy.
- Immunotherapeutic approaches.
- Targeted therapy (FLT3 inhibitors).
- HSCT.
- Second transplant after relapse following a first transplant.
Chemotherapy
Regimens that have been successfully used to induce remission in children with recurrent AML have commonly included high-dose cytarabine given in combination with the following agents:
- Mitoxantrone.[60]
- Fludarabine and idarubicin.[111]
- L-asparaginase.[112]
- Etoposide.
- Liposomal daunorubicin. A study by the International BFM group compared fludarabine, cytarabine, and G-CSF (FLAG) with FLAG plus liposomal daunorubicin. The 4-year OS rate was 38%, with no difference in survival for the total group; however, the addition of liposomal daunorubicin increased the likelihood of obtaining a remission and led to significant improvement in OS in patients with core-binding factor mutations (82%, FLAG plus liposomal daunorubicin vs. 58%, FLAG; P = .04).[113][Level of evidence A1]
- CPX-351. The liposomal combination agent CPX-351, which utilizes a fixed combination of daunorubicin and cytarabine, has been evaluated in the phase I/II COG trial AAML1421 (NCT02642965) for children with relapsed AML. CPX-351 (135 units/m2 /day and containing 60 mg/m2 of daunorubicin) was administered without dexrazoxane in cycle 1 on days 1, 3, and 5 followed by a FLAG cycle. CPX-351 was well tolerated, with no unexpected toxicity, one dose-limiting toxicity (grade 3 ejection fraction decline that resolved), and no toxic mortality. A maculopapular rash occurred in 40% of patients. Among 37 evaluable patients, 75.7% had a CR/CRp/CRi response after the CPX-351 cycle, with 21 of 25 CR/CRp patients having no MRD after cycle 2, and 20 of 25 patients without MRD before HSCT.[114][Level of evidence B4]
- Venetoclax. The SJCRH VENAML trial (NCT03194932) evaluated venetoclax, a selective inhibitor of BCL-2, in combination with cytarabine with or without idarubicin in pediatric patients with relapsed or refractory AML. The combination was well tolerated; the most common grades 3 and 4 adverse events were febrile neutropenia (66% of patients), blood stream infections (16% of patients), and invasive fungal infections (16% of patients). Among the 20 patients treated at the recommended phase II dose, 14 patients (70%) achieved a complete response with or without complete hematological recovery, and 2 patients (10%) achieved a partial response.
Regimens built upon clofarabine and [115,116,117][Level of evidence B4] 2-chloroadenosine have been used.[118] The COG AAML0523 (NCT00372619) trial evaluated the combination of clofarabine plus high-dose cytarabine in patients with relapsed AML; the response rate was 48% and the OS rate, with 21 of 23 responders undergoing HSCT, was 46%. MRD before HSCT was a strong predictor of survival.[119][Level of evidence B4]
The standard-dose cytarabine regimens used in the United Kingdom MRC AML10 study for newly diagnosed children with AML (cytarabine and daunorubicin plus either etoposide or thioguanine) have, when used in the setting of relapse, produced remission rates similar to those achieved with high-dose cytarabine regimens.[104] In a COG phase II study, the addition of bortezomib to idarubicin plus low-dose cytarabine resulted in an overall CR rate of 57%, and the addition of bortezomib to etoposide and high-dose cytarabine resulted in an overall CR rate of 48%.[120]
Immunotherapeutic approaches
Before its FDA approval for use in children with de novo AML in 2020, gemtuzumab ozogamicin was approved for children with relapsed or refractory AML in patients aged 2 years and older.
- The COG AAML00P2 (NCT00028899) study established the maximum tolerated dose (MTD) of gemtuzumab ozogamicin, when combined with mitoxantrone and high-dose cytarabine, as 3 mg/m2; the MTD of gemtuzumab ozogamicin, when combined with Capizzi II based high-dose cytarabine, was 2 mg/m2. [61]
- These regimens produced an overall remission response rate of 45% (±15%), a 1-year EFS rate of 38% (±14%), and a 1-year OS rate of 53% (±15%).
- Sinusoidal occlusion syndrome was seen in one patient with a previous SCT during the cycle containing gemtuzumab ozogamicin and in 4 of 28 patients during subsequent SCT (grade 1 in 2 patients, grade 3 in 1 patient, and grade 4 in 1 patient), all of whom recovered.
- In the dose-escalation portion of the U.K. Medical Research Council AML15 (NCT00006122) study for adults, the dose of gemtuzumab ozogamicin was escalated beyond 3 mg/m2 per dose (the same MTD that was established in the AAML00P2 study) and administered with a conventional intensive chemotherapy backbone; this regimen was not feasible because of hepatotoxicity and delayed hematopoietic recovery. Gemtuzumab ozogamicin at 3 mg/m2 per dose, when given with consecutive courses of intensive chemotherapy, was also not tolerated.[121]
- The Relapsed AML 2001/02 study was a single-arm trial for children (n = 30) who experienced a second relapse or had refractory AML after they did not respond to a second induction regimen. Gemtuzumab ozogamicin as a single agent was dosed at 7.5 mg/dose (children younger than 3 years received 0.25 mg/kg) given every 14 days for two total doses. CR or CRp was seen in 37% of patients; nine patients subsequently underwent SCT, and three of these patients remained in continuous CR. All patients received prophylactic defibrotide during SCT without experiencing any sinusoidal occlusion syndrome (SOS).[122]
- In a prior study of children who received single-agent gemtuzumab ozogamicin, administered at 6 to 9 mg/m2 per dose, patients did not receive defibrotide prophylaxis during subsequent SCT. These studies demonstrated an increased risk of SOS, particularly for patients who underwent SCT less than 3.5 months after the last dose of gemtuzumab ozogamicin.[123]
- Two prospective studies from the Acute Leukemia French Association (ALFA) group examined fractionated gemtuzumab ozogamicin (3 mg/m2 /dose on days 1, 4, and 7) in the relapsed adult AML setting.
- The MYLOFRANCE 1 trial evaluated single-agent fractionated dosing in 57 adults with AML in first relapse, which resulted in a CR rate of 26% and a CRp rate of 7%. No sinusoidal occlusion syndrome occurred during or in subsequent SCT.[124]
- Subsequently, the MYLOFRANCE 2 trial was a phase I/II study (n = 20) that combined the same fractionated dose of gemtuzumab ozogamicin with a dose-finding backbone of daunomycin and cytarabine. Nine patients achieved CR and two patients achieved a CRp. The recommended phase II dose was found to be 60 mg/m2 per day for 3 days for daunomycin and 200 mg/m2 per day for 7 days for cytarabine. No sinusoidal occlusion syndrome was experienced.[125] Fractionated gemtuzumab ozogamicin dosing has been shown to be safe and effective in adults with de novo AML;[27] it is now being evaluated in the MyeChild01 (NCT02724163) phase III study for pediatric patients with de novo AML in the United Kingdom.
Targeted therapy (FLT3 inhibitors)
Midostaurin. There is limited experience with midostaurin in pediatric patients with AML.
- A phase I/II dose-escalation, single-agent trial in 22 children with refractory or relapsed AML (9 with FLT3 mutations) was reported. Seven patients received the initial dose level of 30 mg/m2 given twice daily, and 15 patients received the higher dose level of 60 mg/m2 twice daily, with a median dose duration of 16 days.[35]
- Overall, 72.7% of patients experienced treatment-related adverse events, with only one patient experiencing a dose-limiting toxicity (grades 3–4 ALT elevation).
- In patients with FLT3-mutated AML, 55.5% (21.2%–86.3%) had some clinical response at a median time of 14 days (range, 8–22 days), with one patient achieving a complete remission with incomplete count recovery who was able to proceed to SCT; this patient was the only long-term survivor in this study.
- A phase II trial is under way in Europe, beginning with the 30 mg/m2 twice-daily dosing (NCT03591510).
Gilteritinib. As in de novo AML, most of the focus and published experience with FLT3 inhibitors is in adults with AML and this applies to the relapsed and refractory setting as well. In relapsed or refractory AML, gilteritinib, a type 1 selective FLT3 inhibitor with activity against both FLT3 mutations (ITD and D835/I836 tyrosine kinase domain [TKD]), is the first and only FLT3 inhibitor that has received FDA approval for single-agent use in adults based on the ADMIRAL (NCT02421939) trial.[126]
- In the phase III ADMIRAL trial of adults (aged 18 years and older) with relapsed or refractory FLT3-mutated AML, 247 patients were randomly assigned to receive either single-agent gilteritinib (120 mg/day given once daily) or one of four salvage chemotherapy regimens.[126]
- Median OS was significantly better in patients who received gilteritinib (9.3 months vs. 5.6 months; HR, 0.64; 95% CI, 0.49–0.83; P < .001), with 37.1% versus 16.7% of patients alive at 1 year.
- Importantly, because SCT is felt to be essential for long-term survival in patients with FLT3-mutated AML, a higher percentage of gilteritinib recipients underwent a SCT (25.5% vs. 15.3%). It had equal efficacy in both FLT3 ITD and FLT3 TKD AML cohorts.
- There were fewer adverse events in patients who received gilteritinib than in patients who received salvage chemotherapy regimens; however, some patients who received gilteritinib had elevated hepatic transaminase levels. The main toxic effect was myelosuppression.
Gilteritinib is now being studied in children with FLT3-positive de novo AML in the COG AAML1831 (NCT04293562) trial.
Sorafenib. Sorafenib has been evaluated in pediatric patients with relapsed and refractory AML.
- In a phase I dose de-escalation trial of oral sorafenib in pediatric patients with relapsed or refractory acute leukemia, sorafenib was administered alone on days 1 to 7, and then in combination with clofarabine and high-dose cytarabine for 5 days, followed by single-agent sorafenib use until day 28.[127]
- The recommended phase II dose of sorafenib was determined to be 150 mg/m2 per dose (maximum dose, 300 mg) twice daily (n = 6) after patients experienced significant hand-foot skin reactions (grades 2–3 in 4 of 4 patients; grade 3 dose-limiting toxicities [DLTs] in 2 of 4 patients) at the initial 200 mg/m2 per dose, twice daily level (n = 4).
- Marrow blast reduction was seen in 10 of 12 total patients (4 of 5 patients with FLT3 ITD AML) at day 8.
- Of the 11 patients with AML, 6 patients achieved complete remissions (CR), 2 patients achieved complete remissions with incomplete blood count recovery (CRi), and 1 patient achieved a partial remission (PR) on or after day 22.
- All five patients with FLT3 ITD achieved either CR or CRi.
- A retrospective analysis examined 15 children with AML who received sorafenib for either prophylaxis (n = 6) or relapse (n = 9) after SCT. Doses of sorafenib varied from 75 to 340 mg/m2 per day (median dose, 230 mg/m2) and was given alone in 11 of 15 patients.[128]
- Toxicity was seen in 11 patients, 7 of whom received doses higher than 200 mg/m2; adverse events included count suppression (n = 6), hand-foot skin reactions (n = 6), cardiac dysfunction (n = 2), and others.
- Of the seven patients who experienced DLTs, six patients were able to restart or continue sorafenib treatment after dose adjustments.
- Sorafenib had the greatest efficacy in patients with MRD pre- or post-SCT (five of five patients remained disease free), whereas only one of the six patients who began sorafenib treatment for morphological recurrence remained in CR.
- Graft-versus-host disease was not exacerbated with sorafenib therapy.
HSCT
The selection of additional treatment after the achievement of a second complete remission depends on previous treatment and individual considerations. Consolidation chemotherapy followed by HSCT is conventionally recommended, although there are no controlled prospective data regarding the contribution of additional courses of therapy once a second complete remission is obtained.[102]
Evidence (HSCT after second complete remission):
- The BFM group examined outcomes of children with AML over a 35-year period and found that the greatest improvement in overall outcome was the improvement in survival after relapse. This improved EFS after relapse or refractory disease was only seen in patients who received a SCT as part of their salvage therapy.[129]
- Unrelated donor HSCT has been reported to result in 5-year probabilities of LFS of 45%, 20%, and 12% for patients with AML transplanted in second complete remission, overt relapse, and primary induction failure, respectively.[130][Level of evidence C1]
- A number of studies, including a large, prospective CIBMTR cohort study of children and adults with myeloid diseases, have shown similar or superior survival with busulfan-based regimens compared with TBI.[97,98,131,132]
- Matched-sibling donor transplantation has generally led to the best outcomes, but use of single-antigen mismatched related or matched unrelated donors results in very similar survival at the cost of increased rates of GVHD and nonrelapse mortality.[133] Umbilical cord outcomes are similar to other unrelated donor outcomes, but matching patients at a minimum of 7/8 alleles (HLA A, B, C, DRB1) leads to less nonrelapse mortality.[134] Haploidentical approaches are being used with increasing frequency and have shown comparable outcomes to other stem cell sources in pediatrics.[135] Direct comparison of haploidentical and other unrelated donor sources has not been performed in pediatrics, but studies in adults have shown similar outcomes.[136]
- Reduced-intensity approaches have been used successfully in pediatrics, but mainly in children unable to undergo myeloablative approaches.[137] A randomized trial in adults showed superior outcomes with myeloablative approaches compared with reduced-intensity regimens.[138]
Second transplant after relapse following a first transplant
There is evidence that long-term survival can be achieved in a portion of pediatric patients who undergo a second transplant subsequent to relapse after a first myeloablative transplant. Survival was associated with late relapse (>6–12 months from first transplant), achievement of complete response before the second procedure, and use of a second myeloablative regimen if possible.[139,140,141,142]
CNS relapse
Isolated CNS relapse occurs in 3% to 6% of pediatric AML patients.[66,143,144] Factors associated with an increased risk of isolated CNS relapse include the following:[143]
- Age younger than 2 years at initial diagnosis.
- M5 leukemia.
- 11q23 abnormalities.
- CNS2 or CNS3 involvement at initial diagnosis.[66]
The risk of CNS relapse increases with more CNS leukemic involvement at initial AML diagnosis (CNS1: 0.6%, CNS2: 2.6%, CNS3: 5.8% incidence of isolated CNS relapse, P < .001; multivariate HR for CNS3: 7.82, P = .0003).[66] The outcome of isolated CNS relapse when treated as a systemic relapse is similar to that of bone marrow relapse. In one study, the 8-year OS rate for a cohort of children with an isolated CNS relapse was 26% (± 16%).[143] CNS relapse may also occur in the setting of bone marrow relapse and its likelihood increases with CNS involvement at diagnosis (CNS1: 2.7%, CNS2: 8.5%, CNS3: 9.2% incidence of concurrent CNS relapse, P < .001).[66]
Refractory childhood AML (induction failure)
Treatment options for children with refractory AML may include the following:
- Chemotherapy.
- Gemtuzumab ozogamicin.
Like patients with relapsed AML, induction failure patients are typically directed towards HSCT once they attain a remission, because studies suggest a better EFS than in patients treated with chemotherapy only (31.2% vs. 5%, P < .0001). Attainment of morphological CR for these patients is a significant prognostic factor for DFS after HSCT (46% vs. 0%; P = .02), with failure primarily resulting from relapse (relapse risk, 53.9% vs. 88.9%; P = .02).[145]
Evidence (treatment of refractory childhood AML with gemtuzumab ozogamicin):
- In the SJCRH trial AML02 (NCT00136084), gemtuzumab ozogamicin was given alone (n = 17), typically where MRD was low but still detectable (0.1%–5.6%), or in combination with chemotherapy (n = 29) to those patients with high residual MRD (1%–97%) after the first induction cycle.[146]
- When given alone, 13 of 17 patients became MRD negative.
- When given in combination with chemotherapy, 13 of 29 patients became MRD negative and 28 of 29 patients had reductions in MRD.
- Compared with a nonrandomized cohort of patients with 1% to 25% MRD after induction 1, addition of gemtuzumab ozogamicin to chemotherapy versus chemotherapy alone resulted in significant differences in MRD (P = .03); MRD was eliminated or reduced in all patients who received gemtuzumab ozogamicin versus in only 82% of patients who did not receive gemtuzumab ozogamicin. This was seen despite higher postinduction 1 MRD levels in the cohort of patients who received gemtuzumab ozogamicin (median, 9.5% vs. 2.9% in the no gemtuzumab ozogamicin group, P < .01). There was a nonstatistically significant improvement in 5-year OS rates (55% ± 13.9% vs. 36.4% ± 9.7%, P = .28) and EFS rates (50% ± 9.3% vs. 31.8% ± 13.4%, P = .28).
- No impact on HSCT treatment-related mortality was seen.
- In a phase II trial of gemtuzumab ozogamicin alone for children with relapsed/refractory AML failing previous reinduction attempts, 11 of 30 patients achieved a CR or partial CR, with a 27% versus 0% (P = .001) 3-year OS rate for responders versus nonresponders.[122]
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- AAML1831 (NCT04293562) (A Study to Compare Standard Chemotherapy to Therapy With CPX-351 and/or Gilteritinib for Patients With Newly Diagnosed AML With or Without FLT3 Mutations): This joint industry/COG study is a randomized trial examining whether the liposomal agent CPX-351, which contains daunomycin and cytarabine, improves EFS compared with standard daunomycin and cytarabine. An additional arm for patients with FLT3 ITD AML without favorable cytomolecular characteristics (NPM1 or CEBPA) evaluates the impact of the selective FLT3 inhibitor gilteritinib.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Ries LAG, Melbert D, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2005. National Cancer Institute, 2007. Also available online. Last accessed April 7, 2022.
- Gibson BE, Wheatley K, Hann IM, et al.: Treatment strategy and long-term results in paediatric patients treated in consecutive UK AML trials. Leukemia 19 (12): 2130-8, 2005.
- Lange BJ, Smith FO, Feusner J, et al.: Outcomes in CCG-2961, a children's oncology group phase 3 trial for untreated pediatric acute myeloid leukemia: a report from the children's oncology group. Blood 111 (3): 1044-53, 2008.
- Creutzig U, Büchner T, Sauerland MC, et al.: Significance of age in acute myeloid leukemia patients younger than 30 years: a common analysis of the pediatric trials AML-BFM 93/98 and the adult trials AMLCG 92/99 and AMLSG HD93/98A. Cancer 112 (3): 562-71, 2008.
- Kaspers GJ, Creutzig U: Pediatric acute myeloid leukemia: international progress and future directions. Leukemia 19 (12): 2025-9, 2005.
- Gibson BE, Webb DK, Howman AJ, et al.: Results of a randomized trial in children with Acute Myeloid Leukaemia: medical research council AML12 trial. Br J Haematol 155 (3): 366-76, 2011.
- Creutzig U, Zimmermann M, Lehrnbecher T, et al.: Less toxicity by optimizing chemotherapy, but not by addition of granulocyte colony-stimulating factor in children and adolescents with acute myeloid leukemia: results of AML-BFM 98. J Clin Oncol 24 (27): 4499-506, 2006.
- Cooper TM, Franklin J, Gerbing RB, et al.: AAML03P1, a pilot study of the safety of gemtuzumab ozogamicin in combination with chemotherapy for newly diagnosed childhood acute myeloid leukemia: a report from the Children's Oncology Group. Cancer 118 (3): 761-9, 2012.
- Gamis AS, Alonzo TA, Meshinchi S, et al.: Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children's Oncology Group trial AAML0531. J Clin Oncol 32 (27): 3021-32, 2014.
- Stevens RF, Hann IM, Wheatley K, et al.: Marked improvements in outcome with chemotherapy alone in paediatric acute myeloid leukemia: results of the United Kingdom Medical Research Council's 10th AML trial. MRC Childhood Leukaemia Working Party. Br J Haematol 101 (1): 130-40, 1998.
- Creutzig U, Ritter J, Zimmermann M, et al.: Improved treatment results in high-risk pediatric acute myeloid leukemia patients after intensification with high-dose cytarabine and mitoxantrone: results of Study Acute Myeloid Leukemia-Berlin-Frankfurt-Münster 93. J Clin Oncol 19 (10): 2705-13, 2001.
- Hann IM, Stevens RF, Goldstone AH, et al.: Randomized comparison of DAT versus ADE as induction chemotherapy in children and younger adults with acute myeloid leukemia. Results of the Medical Research Council's 10th AML trial (MRC AML10). Adult and Childhood Leukaemia Working Parties of the Medical Research Council. Blood 89 (7): 2311-8, 1997.
- Burnett AK, Russell NH, Hills RK, et al.: Optimization of chemotherapy for younger patients with acute myeloid leukemia: results of the medical research council AML15 trial. J Clin Oncol 31 (27): 3360-8, 2013.
- Creutzig U, Ritter J, Zimmermann M, et al.: Idarubicin improves blast cell clearance during induction therapy in children with AML: results of study AML-BFM 93. AML-BFM Study Group. Leukemia 15 (3): 348-54, 2001.
- Pession A, Masetti R, Rizzari C, et al.: Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia. Blood 122 (2): 170-8, 2013.
- Burnett AK, Hills RK, Milligan DW, et al.: Attempts to optimize induction and consolidation treatment in acute myeloid leukemia: results of the MRC AML12 trial. J Clin Oncol 28 (4): 586-95, 2010.
- Creutzig U, Zimmermann M, Bourquin JP, et al.: Randomized trial comparing liposomal daunorubicin with idarubicin as induction for pediatric acute myeloid leukemia: results from Study AML-BFM 2004. Blood 122 (1): 37-43, 2013.
- Elgarten CW, Wood AC, Li Y, et al.: Outcomes of intensification of induction chemotherapy for children with high-risk acute myeloid leukemia: A report from the Children's Oncology Group. Pediatr Blood Cancer 68 (12): e29281, 2021.
- Rubnitz JE, Lacayo NJ, Inaba H, et al.: Clofarabine Can Replace Anthracyclines and Etoposide in Remission Induction Therapy for Childhood Acute Myeloid Leukemia: The AML08 Multicenter, Randomized Phase III Trial. J Clin Oncol 37 (23): 2072-2081, 2019.
- Woods WG, Kobrinsky N, Buckley JD, et al.: Timed-sequential induction therapy improves postremission outcome in acute myeloid leukemia: a report from the Children's Cancer Group. Blood 87 (12): 4979-89, 1996.
- Bishop JF, Matthews JP, Young GA, et al.: A randomized study of high-dose cytarabine in induction in acute myeloid leukemia. Blood 87 (5): 1710-7, 1996.
- Becton D, Dahl GV, Ravindranath Y, et al.: Randomized use of cyclosporin A (CsA) to modulate P-glycoprotein in children with AML in remission: Pediatric Oncology Group Study 9421. Blood 107 (4): 1315-24, 2006.
- Rubnitz JE, Inaba H, Dahl G, et al.: Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol 11 (6): 543-52, 2010.
- Pollard JA, Guest E, Alonzo TA, et al.: Gemtuzumab Ozogamicin Improves Event-Free Survival and Reduces Relapse in Pediatric KMT2A-Rearranged AML: Results From the Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 39 (28): 3149-3160, 2021.
- Tarlock K, Alonzo TA, Gerbing RB, et al.: Gemtuzumab Ozogamicin Reduces Relapse Risk in FLT3/ITD Acute Myeloid Leukemia: A Report from the Children's Oncology Group. Clin Cancer Res 22 (8): 1951-7, 2016.
- Guest EM, Aplenc R, Sung L, et al.: Gemtuzumab ozogamicin in infants with AML: results from the Children's Oncology Group trials AAML03P1 and AAML0531. Blood 130 (7): 943-945, 2017.
- Castaigne S, Pautas C, Terré C, et al.: Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 379 (9825): 1508-16, 2012.
- Pollard JA, Loken M, Gerbing RB, et al.: CD33 Expression and Its Association With Gemtuzumab Ozogamicin Response: Results From the Randomized Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 34 (7): 747-55, 2016.
- Olombel G, Guerin E, Guy J, et al.: The level of blast CD33 expression positively impacts the effect of gemtuzumab ozogamicin in patients with acute myeloid leukemia. Blood 127 (17): 2157-60, 2016.
- Lamba JK, Chauhan L, Shin M, et al.: CD33 Splicing Polymorphism Determines Gemtuzumab Ozogamicin Response in De Novo Acute Myeloid Leukemia: Report From Randomized Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 35 (23): 2674-2682, 2017.
- Hills RK, Castaigne S, Appelbaum FR, et al.: Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol 15 (9): 986-96, 2014.
- Perl AE: Availability of FLT3 inhibitors: how do we use them? Blood 134 (9): 741-745, 2019.
- Stone RM, Mandrekar SJ, Sanford BL, et al.: Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 377 (5): 454-464, 2017.
- Schlenk RF, Weber D, Fiedler W, et al.: Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood 133 (8): 840-851, 2019.
- Zwaan CM, Söderhäll S, Brethon B, et al.: A phase 1/2, open-label, dose-escalation study of midostaurin in children with relapsed or refractory acute leukaemia. Br J Haematol 185 (3): 623-627, 2019.
- Pollard JA, Alonzo TA, Gerbing R, et al.: Sorafenib in Combination With Standard Chemotherapy for Children With High Allelic Ratio FLT3/ITD+ Acute Myeloid Leukemia: A Report From the Children's Oncology Group Protocol AAML1031. J Clin Oncol 40 (18): 2023-2035, 2022.
- Sung L, Gamis A, Alonzo TA, et al.: Infections and association with different intensity of chemotherapy in children with acute myeloid leukemia. Cancer 115 (5): 1100-8, 2009.
- Kaya Z, Gursel T, Kocak U, et al.: Invasive fungal infections in pediatric leukemia patients receiving fluconazole prophylaxis. Pediatr Blood Cancer 52 (4): 470-5, 2009.
- Kobayashi R, Kaneda M, Sato T, et al.: The clinical feature of invasive fungal infection in pediatric patients with hematologic and malignant diseases: a 10-year analysis at a single institution at Japan. J Pediatr Hematol Oncol 30 (12): 886-90, 2008.
- Ozer H, Armitage JO, Bennett CL, et al.: 2000 update of recommendations for the use of hematopoietic colony-stimulating factors: evidence-based, clinical practice guidelines. American Society of Clinical Oncology Growth Factors Expert Panel. J Clin Oncol 18 (20): 3558-85, 2000.
- Lehrnbecher T, Zimmermann M, Reinhardt D, et al.: Prophylactic human granulocyte colony-stimulating factor after induction therapy in pediatric acute myeloid leukemia. Blood 109 (3): 936-43, 2007.
- Ehlers S, Herbst C, Zimmermann M, et al.: Granulocyte colony-stimulating factor (G-CSF) treatment of childhood acute myeloid leukemias that overexpress the differentiation-defective G-CSF receptor isoform IV is associated with a higher incidence of relapse. J Clin Oncol 28 (15): 2591-7, 2010.
- Kurt B, Flynn P, Shenep JL, et al.: Prophylactic antibiotics reduce morbidity due to septicemia during intensive treatment for pediatric acute myeloid leukemia. Cancer 113 (2): 376-82, 2008.
- Inaba H, Gaur AH, Cao X, et al.: Feasibility, efficacy, and adverse effects of outpatient antibacterial prophylaxis in children with acute myeloid leukemia. Cancer 120 (13): 1985-92, 2014.
- Sung L, Aplenc R, Alonzo TA, et al.: Effectiveness of supportive care measures to reduce infections in pediatric AML: a report from the Children's Oncology Group. Blood 121 (18): 3573-7, 2013.
- Yeh TC, Liu HC, Hou JY, et al.: Severe infections in children with acute leukemia undergoing intensive chemotherapy can successfully be prevented by ciprofloxacin, voriconazole, or micafungin prophylaxis. Cancer 120 (8): 1255-62, 2014.
- Alexander S, Fisher BT, Gaur AH, et al.: Effect of Levofloxacin Prophylaxis on Bacteremia in Children With Acute Leukemia or Undergoing Hematopoietic Stem Cell Transplantation: A Randomized Clinical Trial. JAMA 320 (10): 995-1004, 2018.
- Taplitz RA, Kennedy EB, Bow EJ, et al.: Antimicrobial Prophylaxis for Adult Patients With Cancer-Related Immunosuppression: ASCO and IDSA Clinical Practice Guideline Update. J Clin Oncol 36 (30): 3043-3054, 2018.
- Ethier MC, Science M, Beyene J, et al.: Mould-active compared with fluconazole prophylaxis to prevent invasive fungal diseases in cancer patients receiving chemotherapy or haematopoietic stem-cell transplantation: a systematic review and meta-analysis of randomised controlled trials. Br J Cancer 106 (10): 1626-37, 2012.
- Robenshtok E, Gafter-Gvili A, Goldberg E, et al.: Antifungal prophylaxis in cancer patients after chemotherapy or hematopoietic stem-cell transplantation: systematic review and meta-analysis. J Clin Oncol 25 (34): 5471-89, 2007.
- Mandhaniya S, Swaroop C, Thulkar S, et al.: Oral voriconazole versus intravenous low dose amphotericin B for primary antifungal prophylaxis in pediatric acute leukemia induction: a prospective, randomized, clinical study. J Pediatr Hematol Oncol 33 (8): e333-41, 2011.
- Mattiuzzi GN, Kantarjian H, Faderl S, et al.: Amphotericin B lipid complex as prophylaxis of invasive fungal infections in patients with acute myelogenous leukemia and myelodysplastic syndrome undergoing induction chemotherapy. Cancer 100 (3): 581-9, 2004.
- Mattiuzzi GN, Kantarjian H, O'Brien S, et al.: Intravenous itraconazole for prophylaxis of systemic fungal infections in patients with acute myelogenous leukemia and high-risk myelodysplastic syndrome undergoing induction chemotherapy. Cancer 100 (3): 568-73, 2004.
- Tacke D, Buchheidt D, Karthaus M, et al.: Primary prophylaxis of invasive fungal infections in patients with haematologic malignancies. 2014 update of the recommendations of the Infectious Diseases Working Party of the German Society for Haematology and Oncology. Ann Hematol 93 (9): 1449-56, 2014.
- Grau S, de la Cámara R, Sabater FJ, et al.: Cost-effectiveness of posaconazole versus fluconazole or itraconazole in the prevention of invasive fungal infections among high-risk neutropenic patients in Spain. BMC Infect Dis 12: 83, 2012.
- Fisher BT, Zaoutis T, Dvorak CC, et al.: Effect of Caspofungin vs Fluconazole Prophylaxis on Invasive Fungal Disease Among Children and Young Adults With Acute Myeloid Leukemia: A Randomized Clinical Trial. JAMA 322 (17): 1673-1681, 2019.
- Getz KD, Sung L, Ky B, et al.: Occurrence of Treatment-Related Cardiotoxicity and Its Impact on Outcomes Among Children Treated in the AAML0531 Clinical Trial: A Report From the Children's Oncology Group. J Clin Oncol 37 (1): 12-21, 2019.
- Feijen EAM, Leisenring WM, Stratton KL, et al.: Derivation of Anthracycline and Anthraquinone Equivalence Ratios to Doxorubicin for Late-Onset Cardiotoxicity. JAMA Oncol 5 (6): 864-871, 2019.
- Getz KD, Sung L, Alonzo TA, et al.: Effect of Dexrazoxane on Left Ventricular Systolic Function and Treatment Outcomes in Patients With Acute Myeloid Leukemia: A Report From the Children's Oncology Group. J Clin Oncol 38 (21): 2398-2406, 2020.
- Wells RJ, Adams MT, Alonzo TA, et al.: Mitoxantrone and cytarabine induction, high-dose cytarabine, and etoposide intensification for pediatric patients with relapsed or refractory acute myeloid leukemia: Children's Cancer Group Study 2951. J Clin Oncol 21 (15): 2940-7, 2003.
- Aplenc R, Alonzo TA, Gerbing RB, et al.: Safety and efficacy of gemtuzumab ozogamicin in combination with chemotherapy for pediatric acute myeloid leukemia: a report from the Children's Oncology Group. J Clin Oncol 26 (14): 2390-3295, 2008.
- Dusenbery KE, Howells WB, Arthur DC, et al.: Extramedullary leukemia in children with newly diagnosed acute myeloid leukemia: a report from the Children's Cancer Group. J Pediatr Hematol Oncol 25 (10): 760-8, 2003.
- Støve HK, Sandahl JD, Abrahamsson J, et al.: Extramedullary leukemia in children with acute myeloid leukemia: A population-based cohort study from the Nordic Society of Pediatric Hematology and Oncology (NOPHO). Pediatr Blood Cancer 64 (12): , 2017.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Superior outcome of pediatric acute myeloid leukemia patients with orbital and CNS myeloid sarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 58 (4): 519-24, 2012.
- Creutzig U, Zimmermann M, Bourquin JP, et al.: CNS irradiation in pediatric acute myleoid leukemia: equal results by 12 or 18 Gy in studies AML-BFM98 and 2004. Pediatr Blood Cancer 57 (6): 986-92, 2011.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Central nervous system disease in pediatric acute myeloid leukemia: A report from the Children's Oncology Group. Pediatr Blood Cancer 64 (12): , 2017.
- Pui CH, Howard SC: Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol 9 (3): 257-68, 2008.
- Pollard JA, Alonzo TA, Brown PA, et al.: Sorafenib in combination with standard chemotherapy for children with high allelic ratio FLT3/ITD+ AML improves event-free survival and reduces relapse risk: a report from the Children's Oncology Group protocol AAML1031. [Abstract] Blood 134 (Suppl 1): A-613, 292, 2019. Also available online. Last accessed April 7, 2022.
- Mayer RJ, Davis RB, Schiffer CA, et al.: Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N Engl J Med 331 (14): 896-903, 1994.
- Wells RJ, Woods WG, Buckley JD, et al.: Treatment of newly diagnosed children and adolescents with acute myeloid leukemia: a Childrens Cancer Group study. J Clin Oncol 12 (11): 2367-77, 1994.
- Wells RJ, Woods WG, Lampkin BC, et al.: Impact of high-dose cytarabine and asparaginase intensification on childhood acute myeloid leukemia: a report from the Childrens Cancer Group. J Clin Oncol 11 (3): 538-45, 1993.
- Getz KD, Alonzo TA, Sung L, et al.: Cytarabine dose reduction in patients with low-risk acute myeloid leukemia: A report from the Children's Oncology Group. Pediatr Blood Cancer 69 (1): e29313, 2022.
- Woods WG, Neudorf S, Gold S, et al.: A comparison of allogeneic bone marrow transplantation, autologous bone marrow transplantation, and aggressive chemotherapy in children with acute myeloid leukemia in remission. Blood 97 (1): 56-62, 2001.
- Horan JT, Alonzo TA, Lyman GH, et al.: Impact of disease risk on efficacy of matched related bone marrow transplantation for pediatric acute myeloid leukemia: the Children's Oncology Group. J Clin Oncol 26 (35): 5797-801, 2008.
- Ravindranath Y, Yeager AM, Chang MN, et al.: Autologous bone marrow transplantation versus intensive consolidation chemotherapy for acute myeloid leukemia in childhood. Pediatric Oncology Group. N Engl J Med 334 (22): 1428-34, 1996.
- Feig SA, Lampkin B, Nesbit ME, et al.: Outcome of BMT during first complete remission of AML: a comparison of two sequential studies by the Children's Cancer Group. Bone Marrow Transplant 12 (1): 65-71, 1993.
- Amadori S, Testi AM, Aricò M, et al.: Prospective comparative study of bone marrow transplantation and postremission chemotherapy for childhood acute myelogenous leukemia. The Associazione Italiana Ematologia ed Oncologia Pediatrica Cooperative Group. J Clin Oncol 11 (6): 1046-54, 1993.
- Bleakley M, Lau L, Shaw PJ, et al.: Bone marrow transplantation for paediatric AML in first remission: a systematic review and meta-analysis. Bone Marrow Transplant 29 (10): 843-52, 2002.
- Koreth J, Schlenk R, Kopecky KJ, et al.: Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 301 (22): 2349-61, 2009.
- Klusmann JH, Reinhardt D, Zimmermann M, et al.: The role of matched sibling donor allogeneic stem cell transplantation in pediatric high-risk acute myeloid leukemia: results from the AML-BFM 98 study. Haematologica 97 (1): 21-9, 2012.
- Creutzig U, Reinhardt D: Current controversies: which patients with acute myeloid leukaemia should receive a bone marrow transplantation?--a European view. Br J Haematol 118 (2): 365-77, 2002.
- Oliansky DM, Rizzo JD, Aplan PD, et al.: The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute myeloid leukemia in children: an evidence-based review. Biol Blood Marrow Transplant 13 (1): 1-25, 2007.
- Niewerth D, Creutzig U, Bierings MB, et al.: A review on allogeneic stem cell transplantation for newly diagnosed pediatric acute myeloid leukemia. Blood 116 (13): 2205-14, 2010.
- Qayed M, Ahn KW, Kitko CL, et al.: A validated pediatric disease risk index for allogeneic hematopoietic cell transplantation. Blood 137 (7): 983-993, 2021.
- Tsukimoto I, Tawa A, Horibe K, et al.: Risk-stratified therapy and the intensive use of cytarabine improves the outcome in childhood acute myeloid leukemia: the AML99 trial from the Japanese Childhood AML Cooperative Study Group. J Clin Oncol 27 (24): 4007-13, 2009.
- Abrahamsson J, Forestier E, Heldrup J, et al.: Response-guided induction therapy in pediatric acute myeloid leukemia with excellent remission rate. J Clin Oncol 29 (3): 310-5, 2011.
- Kelly MJ, Horan JT, Alonzo TA, et al.: Comparable survival for pediatric acute myeloid leukemia with poor-risk cytogenetics following chemotherapy, matched related donor, or unrelated donor transplantation. Pediatr Blood Cancer 61 (2): 269-75, 2014.
- Wareham NE, Heilmann C, Abrahamsson J, et al.: Outcome of poor response paediatric AML using early SCT. Eur J Haematol 90 (3): 187-94, 2013.
- Burke MJ, Wagner JE, Cao Q, et al.: Allogeneic hematopoietic cell transplantation in first remission abrogates poor outcomes associated with high-risk pediatric acute myeloid leukemia. Biol Blood Marrow Transplant 19 (7): 1021-5, 2013.
- Meshinchi S, Alonzo TA, Stirewalt DL, et al.: Clinical implications of FLT3 mutations in pediatric AML. Blood 108 (12): 3654-61, 2006.
- Schlenk RF, Kayser S, Bullinger L, et al.: Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood 124 (23): 3441-9, 2014.
- Beier R, Albert MH, Bader P, et al.: Allo-SCT using BU, CY and melphalan for children with AML in second CR. Bone Marrow Transplant 48 (5): 651-6, 2013.
- Liu DH, Xu LP, Liu KY, et al.: Long-term outcomes of unmanipulated haploidentical HSCT for paediatric patients with acute leukaemia. Bone Marrow Transplant 48 (12): 1519-24, 2013.
- Locatelli F, Masetti R, Rondelli R, et al.: Outcome of children with high-risk acute myeloid leukemia given autologous or allogeneic hematopoietic cell transplantation in the aieop AML-2002/01 study. Bone Marrow Transplant 50 (2): 181-8, 2015.
- Nemecek ER, Hilger RA, Adams A, et al.: Treosulfan, Fludarabine, and Low-Dose Total Body Irradiation for Children and Young Adults with Acute Myeloid Leukemia or Myelodysplastic Syndrome Undergoing Allogeneic Hematopoietic Cell Transplantation: Prospective Phase II Trial of the Pediatric Blood and Marrow Transplant Consortium. Biol Blood Marrow Transplant 24 (8): 1651-1656, 2018.
- Liu H, Zhai X, Song Z, et al.: Busulfan plus fludarabine as a myeloablative conditioning regimen compared with busulfan plus cyclophosphamide for acute myeloid leukemia in first complete remission undergoing allogeneic hematopoietic stem cell transplantation: a prospective and multicenter study. J Hematol Oncol 6: 15, 2013.
- Bredeson C, LeRademacher J, Kato K, et al.: Prospective cohort study comparing intravenous busulfan to total body irradiation in hematopoietic cell transplantation. Blood 122 (24): 3871-8, 2013.
- Dandoy CE, Davies SM, Woo Ahn K, et al.: Comparison of total body irradiation versus non-total body irradiation containing regimens for de novo acute myeloid leukemia in children. Haematologica 106 (7): 1839-1845, 2021.
- Perel Y, Auvrignon A, Leblanc T, et al.: Treatment of childhood acute myeloblastic leukemia: dose intensification improves outcome and maintenance therapy is of no benefit--multicenter studies of the French LAME (Leucémie Aiguë Myéloblastique Enfant) Cooperative Group. Leukemia 19 (12): 2082-9, 2005.
- Arber DA, Vardiman JW, Brunning RD: Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 110-23.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Webb DK: Management of relapsed acute myeloid leukaemia. Br J Haematol 106 (4): 851-9, 1999.
- Stahnke K, Boos J, Bender-Götze C, et al.: Duration of first remission predicts remission rates and long-term survival in children with relapsed acute myelogenous leukemia. Leukemia 12 (10): 1534-8, 1998.
- Webb DK, Wheatley K, Harrison G, et al.: Outcome for children with relapsed acute myeloid leukaemia following initial therapy in the Medical Research Council (MRC) AML 10 trial. MRC Childhood Leukaemia Working Party. Leukemia 13 (1): 25-31, 1999.
- Nakayama H, Tabuchi K, Tawa A, et al.: Outcome of children with relapsed acute myeloid leukemia following initial therapy under the AML99 protocol. Int J Hematol 100 (2): 171-9, 2014.
- Gorman MF, Ji L, Ko RH, et al.: Outcome for children treated for relapsed or refractory acute myelogenous leukemia (rAML): a Therapeutic Advances in Childhood Leukemia (TACL) Consortium study. Pediatr Blood Cancer 55 (3): 421-9, 2010.
- Bachas C, Schuurhuis GJ, Reinhardt D, et al.: Clinical relevance of molecular aberrations in paediatric acute myeloid leukaemia at first relapse. Br J Haematol 166 (6): 902-10, 2014.
- Sander A, Zimmermann M, Dworzak M, et al.: Consequent and intensified relapse therapy improved survival in pediatric AML: results of relapse treatment in 379 patients of three consecutive AML-BFM trials. Leukemia 24 (8): 1422-8, 2010.
- Creutzig U, Zimmermann M, Dworzak MN, et al.: The prognostic significance of early treatment response in pediatric relapsed acute myeloid leukemia: results of the international study Relapsed AML 2001/01. Haematologica 99 (9): 1472-8, 2014.
- Karlsson L, Forestier E, Hasle H, et al.: Outcome after intensive reinduction therapy and allogeneic stem cell transplant in paediatric relapsed acute myeloid leukaemia. Br J Haematol 178 (4): 592-602, 2017.
- Tavil B, Aytac S, Balci YI, et al.: Fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin (FLAG-IDA) for the treatment of children with poor-prognosis acute leukemia: the Hacettepe experience. Pediatr Hematol Oncol 27 (7): 517-28, 2010.
- Capizzi RL, Davis R, Powell B, et al.: Synergy between high-dose cytarabine and asparaginase in the treatment of adults with refractory and relapsed acute myelogenous leukemia--a Cancer and Leukemia Group B Study. J Clin Oncol 6 (3): 499-508, 1988.
- Kaspers GJ, Zimmermann M, Reinhardt D, et al.: Improved outcome in pediatric relapsed acute myeloid leukemia: results of a randomized trial on liposomal daunorubicin by the International BFM Study Group. J Clin Oncol 31 (5): 599-607, 2013.
- Cooper TM, Absalon MJ, Alonzo TA, et al.: Phase I/II Study of CPX-351 Followed by Fludarabine, Cytarabine, and Granulocyte-Colony Stimulating Factor for Children With Relapsed Acute Myeloid Leukemia: A Report From the Children's Oncology Group. J Clin Oncol 38 (19): 2170-2177, 2020.
- Hijiya N, Gaynon P, Barry E, et al.: A multi-center phase I study of clofarabine, etoposide and cyclophosphamide in combination in pediatric patients with refractory or relapsed acute leukemia. Leukemia 23 (12): 2259-64, 2009.
- Jeha S, Razzouk B, Rytting M, et al.: Phase II study of clofarabine in pediatric patients with refractory or relapsed acute myeloid leukemia. J Clin Oncol 27 (26): 4392-7, 2009.
- Shukla N, Kobos R, Renaud T, et al.: Phase II trial of clofarabine with topotecan, vinorelbine, and thiotepa in pediatric patients with relapsed or refractory acute leukemia. Pediatr Blood Cancer 61 (3): 431-5, 2014.
- Chaleff S, Hurwitz CA, Chang M, et al.: Phase II study of 2-chlorodeoxyadenosine plus idarubicin for children with acute myeloid leukaemia in first relapse: a paediatric oncology group study. Br J Haematol 156 (5): 649-55, 2012.
- Cooper TM, Alonzo TA, Gerbing RB, et al.: AAML0523: a report from the Children's Oncology Group on the efficacy of clofarabine in combination with cytarabine in pediatric patients with recurrent acute myeloid leukemia. Cancer 120 (16): 2482-9, 2014.
- Horton TM, Perentesis JP, Gamis AS, et al.: A Phase 2 study of bortezomib combined with either idarubicin/cytarabine or cytarabine/etoposide in children with relapsed, refractory or secondary acute myeloid leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 61 (10): 1754-60, 2014.
- Kell WJ, Burnett AK, Chopra R, et al.: A feasibility study of simultaneous administration of gemtuzumab ozogamicin with intensive chemotherapy in induction and consolidation in younger patients with acute myeloid leukemia. Blood 102 (13): 4277-83, 2003.
- Zwaan CM, Reinhardt D, Zimmerman M, et al.: Salvage treatment for children with refractory first or second relapse of acute myeloid leukaemia with gemtuzumab ozogamicin: results of a phase II study. Br J Haematol 148 (5): 768-76, 2010.
- Arceci RJ, Sande J, Lange B, et al.: Safety and efficacy of gemtuzumab ozogamicin in pediatric patients with advanced CD33+ acute myeloid leukemia. Blood 106 (4): 1183-8, 2005.
- Taksin AL, Legrand O, Raffoux E, et al.: High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia 21 (1): 66-71, 2007.
- Farhat H, Reman O, Raffoux E, et al.: Fractionated doses of gemtuzumab ozogamicin with escalated doses of daunorubicin and cytarabine as first acute myeloid leukemia salvage in patients aged 50-70-year old: a phase 1/2 study of the acute leukemia French association. Am J Hematol 87 (1): 62-5, 2012.
- Perl AE, Martinelli G, Cortes JE, et al.: Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med 381 (18): 1728-1740, 2019.
- Inaba H, Rubnitz JE, Coustan-Smith E, et al.: Phase I pharmacokinetic and pharmacodynamic study of the multikinase inhibitor sorafenib in combination with clofarabine and cytarabine in pediatric relapsed/refractory leukemia. J Clin Oncol 29 (24): 3293-300, 2011.
- Tarlock K, Chang B, Cooper T, et al.: Sorafenib treatment following hematopoietic stem cell transplant in pediatric FLT3/ITD acute myeloid leukemia. Pediatr Blood Cancer 62 (6): 1048-54, 2015.
- Rasche M, Zimmermann M, Borschel L, et al.: Successes and challenges in the treatment of pediatric acute myeloid leukemia: a retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 32 (10): 2167-2177, 2018.
- Bunin NJ, Davies SM, Aplenc R, et al.: Unrelated donor bone marrow transplantation for children with acute myeloid leukemia beyond first remission or refractory to chemotherapy. J Clin Oncol 26 (26): 4326-32, 2008.
- Woodard P, Carpenter PA, Davies SM, et al.: Unrelated donor bone marrow transplantation for myelodysplastic syndrome in children. Biol Blood Marrow Transplant 17 (5): 723-8, 2011.
- Uberti JP, Agovi MA, Tarima S, et al.: Comparative analysis of BU and CY versus CY and TBI in full intensity unrelated marrow donor transplantation for AML, CML and myelodysplasia. Bone Marrow Transplant 46 (1): 34-43, 2011.
- Shaw PJ, Kan F, Woo Ahn K, et al.: Outcomes of pediatric bone marrow transplantation for leukemia and myelodysplasia using matched sibling, mismatched related, or matched unrelated donors. Blood 116 (19): 4007-15, 2010.
- Eapen M, Klein JP, Ruggeri A, et al.: Impact of allele-level HLA matching on outcomes after myeloablative single unit umbilical cord blood transplantation for hematologic malignancy. Blood 123 (1): 133-40, 2014.
- Locatelli F, Merli P, Pagliara D, et al.: Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood 130 (5): 677-685, 2017.
- Rashidi A, DiPersio JF, Westervelt P, et al.: Comparison of Outcomes after Peripheral Blood Haploidentical versus Matched Unrelated Donor Allogeneic Hematopoietic Cell Transplantation in Patients with Acute Myeloid Leukemia: A Retrospective Single-Center Review. Biol Blood Marrow Transplant 22 (9): 1696-1701, 2016.
- Pulsipher MA, Boucher KM, Wall D, et al.: Reduced-intensity allogeneic transplantation in pediatric patients ineligible for myeloablative therapy: results of the Pediatric Blood and Marrow Transplant Consortium Study ONC0313. Blood 114 (7): 1429-36, 2009.
- Scott BL, Pasquini MC, Logan BR, et al.: Myeloablative Versus Reduced-Intensity Hematopoietic Cell Transplantation for Acute Myeloid Leukemia and Myelodysplastic Syndromes. J Clin Oncol 35 (11): 1154-1161, 2017.
- Meshinchi S, Leisenring WM, Carpenter PA, et al.: Survival after second hematopoietic stem cell transplantation for recurrent pediatric acute myeloid leukemia. Biol Blood Marrow Transplant 9 (11): 706-13, 2003.
- Nishikawa T, Inagaki J, Nagatoshi Y, et al.: The second therapeutic trial for children with hematological malignancies who relapsed after their first allogeneic SCT: long-term outcomes. Pediatr Transplant 16 (7): 722-8, 2012.
- Yaniv I, Krauss AC, Beohou E, et al.: Second Hematopoietic Stem Cell Transplantation for Post-Transplantation Relapsed Acute Leukemia in Children: A Retrospective EBMT-PDWP Study. Biol Blood Marrow Transplant 24 (8): 1629-1642, 2018.
- Uden T, Bertaina A, Abrahamsson J, et al.: Outcome of children relapsing after first allogeneic haematopoietic stem cell transplantation for acute myeloid leukaemia: a retrospective I-BFM analysis of 333 children. Br J Haematol 189 (4): 745-750, 2020.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Risk factors and therapy for isolated central nervous system relapse of pediatric acute myeloid leukemia. J Clin Oncol 23 (36): 9172-8, 2005.
- Abbott BL, Rubnitz JE, Tong X, et al.: Clinical significance of central nervous system involvement at diagnosis of pediatric acute myeloid leukemia: a single institution's experience. Leukemia 17 (11): 2090-6, 2003.
- Quarello P, Fagioli F, Basso G, et al.: Outcome of children with acute myeloid leukaemia (AML) experiencing primary induction failure in the AIEOP AML 2002/01 clinical trial. Br J Haematol 171 (4): 566-73, 2015.
- O'Hear C, Inaba H, Pounds S, et al.: Gemtuzumab ozogamicin can reduce minimal residual disease in patients with childhood acute myeloid leukemia. Cancer 119 (22): 4036-43, 2013.
Acute Promyelocytic Leukemia (APL)
Acute promyelocytic leukemia (APL) is a distinct subtype of acute myeloid leukemia (AML) because of several factors, including the following:
- Clinical presentation of universal coagulopathy (disseminated intravascular coagulation) and unique morphological characteristics (French-American-British [FAB] M3 or its variants).
- Unique molecular etiology as a result of the involvement of the RARA oncogene.
- Unique sensitivity to the differentiating agent tretinoin and to the proapoptotic agent arsenic trioxide.[1]
These unique features of APL mandate a high index of suspicion at diagnosis so as to initiate proper supportive care measures to avoid coagulopathic complications during the first days of therapy. It is also critical to institute a different induction regimen of therapy to minimize the risk of coagulopathic complications and to provide a much improved long-term relapse-free survival and overall survival (OS) than with past approaches to treating APL and compared with outcomes for patients with the other forms of AML.[2,3]
Molecular Abnormality
The characteristic chromosomal abnormality associated with APL is t(15;17). This translocation involves a breakpoint that includes the retinoic acid receptor and leads to production of the promyelocytic leukemia (PML)::retinoic acid receptor alpha (RARA) fusion protein.[1]
Patients with a suspected diagnosis of APL can have their diagnosis confirmed by detection of the PML::RARA fusion protein (e.g., through fluorescence in situ hybridization [FISH], reverse transcriptase–polymerase chain reaction [RT-PCR], or conventional cytogenetics). An immunofluorescence method using an anti-PML monoclonal antibody can rapidly establish the presence of the PML::RARA fusion protein based on the characteristic distribution pattern of PML that occurs in the presence of the fusion protein.[4,5,6]
Clinical Presentation
Clinically, APL is characterized by severe coagulopathy that is often present at the time of diagnosis.[7] This is typically manifested with thrombocytopenia, prolonged prothrombin time, partial thromboplastin time, elevated d-dimers, and hypofibrinogenemia.[8] Mortality during induction (particularly with cytotoxic agents used alone) caused by bleeding complications is more common in this subtype than in other FAB or World Health Organization (WHO) classifications.[9,10] A multicooperative group analysis of children with APL who were treated with tretinoin and chemotherapy reported that early induction coagulopathic deaths occurred in 25 of 683 children (3.7%); 23 deaths resulted from hemorrhage (19 CNS, 4 pulmonary), and 2 resulted from CNS thrombosis.[11] A lumbar puncture at diagnosis should not be performed until evidence of coagulopathy has resolved.
Tretinoin therapy is initiated as soon as APL is suspected on the basis of morphological and clinical presentation,[2,12] because tretinoin has been shown to ameliorate bleeding risk for patients with APL.[13] A retrospective analysis identified an increase in early death resulting from hemorrhage in patients with APL in whom tretinoin introduction was delayed.[8] This emergent need led to the early administration of tretinoin while not precluding participation in other AML clinical trials if the APL diagnosis proves to be incorrect. Additionally, initiation of supportive measures such as replacement transfusions directed at correction of the coagulopathy is critical during these initial days of diagnosis and therapy. Patients at greatest risk of coagulopathic complications are those presenting with high white blood cell (WBC) counts, high body mass index, hypofibrinogenemia, molecular variants of APL, and the presence of FLT3 internal tandem duplication (ITD) mutations.[8,11]
APL in children is generally similar to APL in adults, although children have a higher incidence of hyperleukocytosis (defined as WBC count higher than 10 × 109 /L) and a higher incidence of the microgranular morphological subtype.[14,15,16,17] As in adults, children with WBC counts less than 10 × 109 /L at diagnosis have significantly better outcomes than do patients with higher WBC counts.[15,16,18]
Risk Classification for Treatment Stratification
The prognostic significance of WBC count is used to define high-risk and low-risk patient populations and to assign postinduction treatment, with high-risk patients most commonly defined by WBC count of 10 × 109 /L or greater.[19,20]FLT3 mutations (either ITD or tyrosine kinase domain [TKD] mutations) are observed in 40% to 50% of APL cases, with the presence of FLT3 mutations correlating with higher WBC counts and the microgranular variant (M3v) subtype.[21,22,23,24,25] The FLT3 mutation has been associated with an increased risk of induction death and, in some reports, an increased risk of treatment failure.[21,22,23,24,25,26,27]
In the COG AAML0631 (NCT00866918) trial, which included treatment with chemotherapy, tretinoin, and arsenic trioxide, risk classification primarily defined early death risk rather than relapse risk (standard risk, 0 of 66 patients vs. high risk, 4 of 35 patients).[28] In the COG AAML0631 (NCT00866918) and AAML1331 (NCT02339740) trials, relapse risk after remission induction was 4% and 2% overall, respectively.[28,29] High-risk patients in these trials had earlier initiation of idarubicin, with the first dose on day 1 rather than day 3 to reduce leukemic burden more rapidly.[28]
The Central Nervous System (CNS) and APL
CNS involvement at the time of diagnosis is not ascertained in most patients with APL because of the presence of disseminated intravascular coagulation. The COG AAML0631 (NCT00866918) trial identified 28 patients out of 101 enrolled children who had cerebrospinal fluid (CSF) exams at diagnosis, and in 7 of these children, blasts were identified in atraumatic taps.[28] None of the patients experienced a CNS relapse with intrathecal treatment during induction and prophylactic doses during therapy.
Overall, CNS relapse is uncommon for patients with APL, particularly for those with WBC counts of less than 10 × 109 /L.[30,31] In two clinical trials enrolling more than 1,400 adults with APL in which CNS prophylaxis was not administered, the cumulative incidence of CNS relapse was less than 1% for patients with WBC counts of less than 10 × 109 /L, while it was approximately 5% for those with WBC counts of 10 × 109 /L or greater.[30,31] In addition to high WBC counts at diagnosis, CNS hemorrhage during induction is also a risk factor for CNS relapse.[31] A review of published cases of pediatric APL also observed low rates of CNS relapse. Because of the low incidence of CNS relapse among children with APL presenting with WBC counts of less than 10 × 109 /L, CNS surveillance and prophylactic CNS therapy may not be needed for this group of patients,[32] although there is no consensus on this topic.[33]
Treatment of APL
Modern treatment programs for APL are based on the sensitivity of leukemia cells from APL patients to the differentiation-inducing and apoptotic effects of tretinoin and arsenic trioxide. APL therapy first diverged from the therapy of other non-APL subtypes of AML with the addition of tretinoin to chemotherapy. With the incorporation of arsenic trioxide into modern treatment regimens, the use of traditional chemotherapy in adults and children is increasingly restricted to the induction phase for high-risk patients.[29,34,35]
Treatment options for children with APL may include the following:
- Tretinoin.
- Arsenic trioxide.
- Chemotherapy.
- Supportive care.
Given the very high level of activity for the combination of arsenic trioxide and tretinoin for adults with APL,[34,35] and given data indicating that children with APL have a similar response to these agents,[29,36,37,38,39] the use of these two agents alone in standard-risk patients, and with short-course chemotherapy during induction in high-risk patients, is the optimal therapeutic approach for this disease.
Before this approach was discovered, chemotherapy was used in all or most phases of therapy including induction, consolidation, and maintenance for pediatric trials like AAML0631 (NCT00866918). The regimens that use chemotherapy are now primarily of historical interest. They can also be used as a reference in refractory cases because of the findings from randomized clinical trials that compared regimens with the combination of tretinoin and arsenic trioxide with or without chemotherapy. Results from the completed cooperative group trial (COG AAML1331 [NCT02339740]) verified the benefit of treatment with tretinoin and arsenic trioxide for children with newly diagnosed APL,[29] similar to results reported by other groups.[39] The dramatic efficacy of tretinoin against APL results from the ability of pharmacological doses of tretinoin to overcome the repression of signaling caused by the PML::RARA fusion protein at physiological tretinoin concentrations. Restoration of signaling leads to differentiation of APL cells and then to postmaturation apoptosis.[40] Most patients with APL achieve a complete remission (CR) when treated with tretinoin, although single-agent tretinoin is generally not curative.[41,42]
Almost all children with APL who were treated with tretinoin, arsenic trioxide, and modern supportive care (outlined below) achieved CR in the absence of coagulopathy-related mortality.[15,16,29,43,44,45]
Assessment of response to induction therapy in the first month of treatment using morphological and molecular criteria may provide misleading results because delayed persistence of differentiating leukemia cells can occur in patients who will ultimately achieve CR.[2,3] Alterations in planned treatment based on these early observations are not appropriate because resistance of APL to tretinoin plus arsenic trioxide is rare.[20,29,46]
For patients with APL, consolidation therapy may include repeated cycles of tretinoin and arsenic trioxide without additional chemotherapy, based on the adult and pediatric experience.[29,34,35,39] Studies using arsenic trioxide–based consolidation have demonstrated excellent survival without cytarabine consolidation.[26,29,34,39,47]
Maintenance therapy is likely unnecessary for patients with APL who are treated with tretinoin and arsenic trioxide based on data from adult trials and the COG AAML1331 (NCT02339740) trial.[29] Because of the favorable outcomes with tretinoin and arsenic trioxide, hematopoietic stem cell transplantation is not recommended in first CR.
Arsenic trioxide is the most active agent in the treatment of APL, and while initially used in relapsed APL, it is now incorporated into the treatment of newly diagnosed patients. Data supporting the use of arsenic trioxide initially came from trials that included adult patients only, but more recently, its efficacy has been seen in trials that included pediatric patients.
Evidence (arsenic trioxide therapy):
- In adults with newly diagnosed APL treated on the CALGB-C9710 (NCT00003934) trial, the addition of two consolidation courses of arsenic trioxide to a standard APL treatment regimen resulted in the following:[47]
- A significant improvement in EFS (80% vs. 63% at 3 years; P < .0001) and disease-free survival (DFS) rates (90% vs. 70% at 3 years; P < .0001), although the outcome of patients who did not receive arsenic trioxide was inferior to the results obtained in the Gruppo Italiano Malattie EMatologiche dell'Adulto (GIMEMA) or PETHEMA trials.
- In children and adolescents with newly diagnosed APL treated on the COG AAML0631 (NCT00866918) trial, two consolidation cycles of arsenic trioxide were incorporated into a chemotherapy regimen with lower cumulative anthracycline doses compared with historical controls.[28]
- The 3-year OS rate was 94%, and the EFS rate was 91%.
- Patients with standard-risk APL had an OS rate of 98% and an EFS rate of 95%.
- High-risk patients had an OS rate of 86% and an EFS rate of 83%. This lower survival compared with standard-risk patients was primarily caused by early death events.
- The relapse risk after arsenic trioxide consolidation was 4% and was similar for standard-risk and high-risk APL.
- The concurrent use of arsenic trioxide and tretinoin in newly diagnosed patients with APL results in high rates of CR.[48,49,50] Early experience in children with newly diagnosed APL showed high rates of CR to arsenic trioxide, either as a single agent or given with tretinoin.[51][Level of evidence C1] Results of a meta-analysis of seven published studies in adult patients with APL suggested that using a combination of arsenic trioxide and tretinoin may be more effective than using arsenic trioxide alone to induce CR.[52]
- Arsenic trioxide was evaluated as a component of induction therapy with idarubicin and tretinoin in the APML4 clinical trial, which enrolled both children and adults (N = 124 evaluable patients).[26] Patients received two courses of consolidation therapy with arsenic trioxide and tretinoin (but no anthracycline) and maintenance therapy with tretinoin, mercaptopurine, and methotrexate.[55]
- The 2-year freedom-from-relapse rate was 97.5%, the failure-free survival (FFS) rate was 88.1%, and the OS rate was 93.2%.
- These outcome results are superior to those reported for patients who did not receive arsenic trioxide in the predecessor clinical trial (APML3).
- A German and Italian phase III clinical trial (APL0406 [NCT00482833]) compared tretinoin plus chemotherapy with tretinoin plus arsenic trioxide in adults with APL classified as low to intermediate risk (WBC ≤10 × 109 /L).[34] Patients were randomly assigned to receive either tretinoin plus arsenic trioxide for induction and consolidation therapy or standard tretinoin-idarubicin induction therapy followed by three cycles of consolidation therapy with tretinoin plus chemotherapy and maintenance therapy with low-dose chemotherapy and tretinoin.
- All patients who received tretinoin plus arsenic trioxide (n = 77) achieved CR at the end of induction therapy, while 95% of patients who received tretinoin plus chemotherapy (n = 79) achieved CR.
- EFS rates were 97% in the tretinoin-arsenic trioxide group compared with 86% in the tretinoin-chemotherapy group (P = .02).
- Two-year OS probability was 99% (95% confidence interval [CI], 96%–100%) in the tretinoin-arsenic trioxide group and 91% (95% CI, 85%–97%) in the tretinoin-chemotherapy group (P = .02).
- An updated longer-term analysis demonstrated that at 50 months, the tretinoin-arsenic trioxide arm showed even greater superiority, with OS rates of 97% versus 80% (P < .001).[34,35]
- These results indicate that low-risk to intermediate-risk APL is curable for a high percentage of patients without conventional chemotherapy.
- The historically controlled noninferiority COG AAML1331 (NCT02339740) trial was conducted between 2015 and 2019. The study included pediatric patients (age range, 1–21 years) with APL. The study examined whether the addition of arsenic trioxide to induction therapy, and continued through consolidation, could sustain the excellent outcomes seen in the AAML0631 (NCT00866918) trial. Additionally, chemotherapy was eliminated entirely, except when patients with high-risk APL were given short courses of idarubicin during induction therapy. Patients with standard-risk APL had idarubicin eliminated from the induction cycle. Mitoxantrone, high-dose cytarabine, and idarubicin were eliminated from the consolidation cycles. Then, mercaptopurine and methotrexate were eliminated from the maintenance cycles. Intrathecal doses of cytarabine were also eliminated. The AAML1331 study included 158 patients, 98 of whom were classified as standard risk and 56 of whom were classified as high risk.[29]
Standard-risk patients received tretinoin plus arsenic trioxide on days 1 to 28, with the possibility of continuing treatment up to day 70 to achieve a CR. High-risk patients received the same induction therapy schedule as standard-risk patients, with the addition of idarubicin on induction days 1, 3, 5, and 7. High-risk patients also received daily dexamethasone as a prophylactic treatment to prevent differentiation syndrome on days 1 to 14. All patients received the same consolidation therapy, which consisted of tretinoin on days 1 to 14 and days 29 to 42. Patients were also given arsenic trioxide 5 days each week for 4 consecutive weeks in every 8-week cycle (three rounds). The fourth consolidation therapy cycle concluded on day 28. There was no maintenance therapy phase.
- The median duration of induction therapy for all patients (standard risk and high risk) was 47 days. Induction therapy included a 14-day rest period before starting consolidation therapy. All standard-risk and high-risk patients who completed their induction therapy achieved a hematologic CR or a CR with incomplete hematologic recovery before day 70.
- During induction therapy, one standard-risk patient died of complications from coagulopathy, differentiation syndrome, and subsequent organ failure. No high-risk patients died of complications.
- All patients who received quantitative PCR testing after completing their second round of consolidation therapy were in molecular remission.
- No patients experienced a relapse while on therapy. One standard-risk patient (1%, CNS recurrence) and two high-risk patients (4%) experienced relapses after therapy completion. These patients were successfully salvaged.
- The AAML1331 and AAML0631 trials were compared and the following was reported:
- Standard-risk patients had equivalent 2-year EFS rates (98% vs. 97%) and OS rates (99% vs. 98.5%).
- High-risk patients who enrolled in the AAML1331 trial had a significantly improved 2-year EFS rate (96.4% vs. 82.9%, P = 0.05) and OS rate (100% vs. 85.7%, P = .02).
- In the AAML1331 trial, patients with CNS symptoms or hemorrhage were examined and treated using triple intrathecal chemotherapy, whereas in the AAML0631 study, standard-risk patients received three prophylactic doses of intrathecal chemotherapy, and high-risk patients received four prophylactic doses of intrathecal chemotherapy.
- The length of therapy was significantly shorter in the AAML1331 trial (9 months) than in the AAML0631 trial (>2 years).
- Hospitalizations during consolidation therapy were significantly reduced in the AAML1331 trial, when compared with the AAML0631 trial (0 days vs. 13 days, respectively; P < .001).
- In the AAML1331 trial, early death was significantly lower in high-risk patients (0 vs. 4 in AAML0631, P = .02), and not significantly different for standard-risk patients (1 vs. 0 in AAML0631, P = .16).
In summary, survival rates for children with APL exceeding 90% are achievable using treatment programs that prescribe the rapid initiation of tretinoin with appropriate supportive care measures and that combine arsenic trioxide with tretinoin for induction and consolidation therapy.[29,39] Cytotoxic chemotherapy is required only for high-risk patients, and its use is restricted to induction therapy.[29] For patients in CR for more than 5 years, relapse is extremely rare.[56][Level of evidence B1]
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Complications unique to APL therapy
In addition to the previously mentioned universal presence of coagulopathy in patients newly diagnosed with APL (further described below), several other unique complications occur in patients with APL for which the clinician should be aware. These include two tretinoin-related conditions, pseudotumor cerebri and differentiation syndrome (also called retinoic acid syndrome), and an arsenic trioxide–related complication, QT interval prolongation.
- Pseudotumor cerebri. Pseudotumor cerebri is typically manifested by headache, papilledema, sixth nerve palsy, visual field cuts, and normal intracranial imaging in the face of an elevated opening lumbar puncture pressure (not often obtained in APL patients). Pseudotumor cerebri is known to be associated with tretinoin, presumably by the same mechanism of vitamin A toxicity that leads to increased production of cerebrospinal fluid.
The incidence of pseudotumor cerebri has been reported to be as low as 1.7% with very strict definitions of the complication and as high as 6% to 16% in pediatric trials.[14,15,28,29,57] Pseudotumor cerebri is thought to be more prevalent in children receiving tretinoin, leading to lower dosing in contemporary pediatric APL clinical trials.[14,29] Pseudotumor cerebri most typically occurs during induction at a median of 15 days (range, 1–35 days) after starting tretinoin, but is known to occur in other phases of therapy as well.[57] Pseudotumor cerebri incidence and severity may be exacerbated with the concurrent use of azoles via inhibition of cytochrome P450 metabolism of tretinoin.
When a diagnosis of pseudotumor cerebri is suspected, tretinoin is with held until symptoms abate and then is slowly escalated to full dose as tolerated.[57]
- Differentiation syndrome. Differentiation syndrome (also known as retinoic acid syndrome or tretinoin syndrome) is a life-threatening syndrome thought to be an inflammatory response–mediated syndrome manifested by weight gain, fever, edema, pulmonary infiltrates, pleuro-pericardial effusions, hypotension, and, in the most severe cases, acute renal failure.[58] In the contemporary COG AAML0631 (NCT00866918) study, it was present in 20% of patients during induction and was more prevalent in high-risk children (31%) than in low-risk children (13%), a risk factor also seen in adults with APL.[28,59] There is a bimodal peak with this syndrome seen in the first and third weeks of induction therapy.
Since differentiation syndrome occurs more often in high-risk patients, dexamethasone is given with tretinoin and/or arsenic trioxide to prevent this complication.[58] Prophylaxis with dexamethasone and hydroxyurea (for cytoreduction) is also administered to standard-risk patients if their WBC count rises to greater than 10,000/µL after the start of tretinoin or arsenic. If differentiation syndrome occurs, the patient's dexamethasone dose may be escalated with temporary with holding of tretinoin and arsenic trioxide and, similar to pseudotumor cerebri, restarted at a lower dose and escalated as tolerated. When this approach was used in the COG AAML1331 (NCT02339740) trial, 24.5% of standard-risk patients and 30.4% of high-risk patients presented with differentiation syndrome. Only one standard-risk patient died from differentiation syndrome and coagulopathy.[29]
- Coagulopathy. Along with differentiation syndrome, coagulopathy complications result in a higher risk of death during induction (early death in APL). APL blasts induce coagulopathy by activation of the coagulation cascade (caused by the expression of tissue factor and other procoagulants) with concomitant increase in primary and secondary fibrinolysis resulting from expression of annexin II on the APL blasts. Risk of death caused by coagulopathy is associated with an elevated WBC count, decreased platelet count, and abnormal coagulation studies (fibrinogen, prothrombin time).[12] Studies of both adult and pediatric patients have demonstrated that scoring systems using clinical characteristics and laboratory values can help predict the risk of developing severe or lethal coagulopathy.[60,61] Aggressive supportive care to correct coagulopathy, even before clinical signs and symptoms of bleeding or thrombosis occur, is important to prevent early death.
- QT interval prolongation. Arsenic trioxide is associated with QT interval prolongation that can lead to life-threatening arrhythmias (e.g., torsades de pointes).[62] It is essential to monitor electrolytes closely in patients receiving arsenic trioxide and to maintain potassium and magnesium values at midnormal ranges, as well as to be cognizant of other agents known to prolong the QT interval.[63]
Minimal disease monitoring
The induction and consolidation therapies currently employed result in molecular remission, as measured by RT-PCR for PML::RARA fusion protein, in most APL patients, with 1% or fewer showing molecular evidence of disease at the end of consolidation therapy.[20,46] While two negative RT-PCR assays after completion of therapy are associated with long-term remission,[64] conversion from negative to positive RT-PCR is highly predictive of subsequent hematologic relapse.[65]
Patients with persistent or relapsing disease on the basis of PML::RARA fusion protein RT-PCR measurement may benefit from intervention with relapse therapies [66,67]. For more information, see the Treatment of Recurrent APL section.
Molecular Variants of APL Other ThanPML::RARAand Therapeutic Impact
Uncommon molecular variants of APL produce fusion proteins that join distinctive gene partners (e.g., ZBTB16, NPM1, STAT5B, and NUMA1) to RARA.[68,69] Recognition of these rare variants is important because they differ in their sensitivity to tretinoin and to arsenic trioxide.[70]
- ZBTB16::RARA fusion variant. The ZBTB16::RARA fusion variant, characterized by t(11;17)(q23;q21), represents about 0.8% of APL, expresses surface CD56, and has very fine granules compared with t(15;17) APL.[71,72,73] Patients with APL and ZBTB16::RARA fusions have a poor prognosis and do not usually respond to tretinoin or arsenic trioxide.[70,71,72,73]
- NPM1::RARA or NUMA1::RARA fusion variant. The rare APL variants with NPM1::RARA fusions (t(5;17)(q35;q21)) or NUMA1::RARA fusions (t(11;17)(q13;q21)) may still be responsive to tretinoin.[70,74,75,76,77]
Treatment of Recurrent APL
Historically, 10% to 20% of patients with APL relapsed; however, current studies that incorporated arsenic trioxide therapy showed a cumulative incidence of relapse of less than 5%.[28,29,35]
In patients who initially received chemotherapy-based treatments, the duration of their first remission was prognostic in APL, with patients who relapsed within 12 to 18 months of initial diagnosis having a worse outcome.[78,79,80]
Many children with APL who relapsed were exposed to anthracyclines in previous trials (exposures ranged from 400 mg/m2 to 750 mg/m2).[2] Thus, regimens containing anthracyclines were often not optimal for children with APL who relapsed.
Treatment options for children with recurrent APL may include the following:
- Arsenic trioxide or tretinoin.
- Gemtuzumab ozogamicin.
- Hematopoietic stem cell transplantation (HSCT).
Arsenic trioxide
For children with recurrent APL, the use of arsenic trioxide as a single agent or in regimens including tretinoin should be considered, depending on the therapy given during first remission. Arsenic trioxide is an active agent in patients with recurrent APL, with approximately 85% to 94% of patients achieving remission after treatment with this agent.[48,81,82,83,84,85] Arsenic trioxide is even capable of inducing remissions in patients who relapse after having received arsenic trioxide with or without other agents during initial therapy.[85,86] APL cells, however, may become resistant to arsenic trioxide through mechanisms including mutation of the PML domain of the PML::RARA fusion oncogene.[87]
For adults with relapsed APL, approximately 85% to 94% achieve morphological remission after treatment with arsenic trioxide.[82,83,85,88] Data, although more limited in children, suggest that children with relapsed APL have a response to arsenic trioxide similar to that of adults.[81,83,85,89] Arsenic trioxide is well tolerated in children with relapsed APL. The toxicity profile and response rates in children are similar to that observed in adults.[81,85]
Because arsenic trioxide causes QT-interval prolongation that can lead to life-threatening arrhythmias,[62] it is essential to monitor electrolytes closely in patients receiving arsenic trioxide and to maintain potassium and magnesium values at midnormal ranges.[63]
Gemtuzumab ozogamicin
The use of gemtuzumab ozogamicin, an anti-CD33/calicheamicin monoclonal antibody, as a single agent resulted in a 91% (9 of 11 patients) molecular remission after two doses and a 100% (13 of 13 patients) molecular remission after three doses, thus, demonstrating excellent activity of this agent in patients with relapsed APL.[90]
HSCT
Retrospective pediatric studies have reported 5-year EFS rates after either autologous or allogeneic transplantation approaches to be similar, at approximately 70%.[91,92]
Evidence (autologous HSCT):
- When considering autologous transplantation, a study in adult patients demonstrated improved 7-year EFS rates (77% vs. 50%) when both the patient and the stem cell product had negative promyelocytic leukemia/retinoic acid receptor alpha fusion transcript by polymerase chain reaction (molecular remission) before transplant.[93]
- Another study demonstrated that among seven patients undergoing autologous HSCT and whose cells were MRD positive, all relapsed in less than 9 months after transplantation; however, only one of eight patients whose autologous donor cells were MRD negative relapsed.[94]
- Another report demonstrated that the 5-year EFS rate was 83.3% for patients who underwent autologous HSCT in second molecular remission and was 34.5% for patients who received only maintenance therapy.[95]
- Another retrospective report found that 94% of pediatric and adult patients (64 of 67) with relapsed APL, after primarily receiving single-agent arsenic trioxide, achieved a molecular remission after treatment with arsenic-containing reinduction regimens. For patients (n = 35) who received postremission consolidation with HSCT, the 5-year OS rate was 90.3% (± 5.3%), and the EFS rate was 87.1% (± 6.0%). These outcomes were significantly superior to the outcomes of patients who received an arsenic-containing maintenance regimen (n = 28); these patients had a 5-year OS rate of 58.6% (± 10.4%) and an EFS rate of 47.7% (± 10.3%).[85]
Such data support the use of autologous transplantation in patients who are MRD negative in second CR and have MRD-negative stem cell collections.
Because of the rarity of APL in children and the favorable outcome for this disease, clinical trials in relapsed APL to compare treatment approaches are likely not feasible. However, an international expert panel provided recommendations for the treatment of relapsed APL on the basis of the reported pediatric and adult experience.[96]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Melnick A, Licht JD: Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93 (10): 3167-215, 1999.
- Sanz MA, Grimwade D, Tallman MS, et al.: Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 113 (9): 1875-91, 2009.
- Sanz MA, Lo-Coco F: Modern approaches to treating acute promyelocytic leukemia. J Clin Oncol 29 (5): 495-503, 2011.
- Falini B, Flenghi L, Fagioli M, et al.: Immunocytochemical diagnosis of acute promyelocytic leukemia (M3) with the monoclonal antibody PG-M3 (anti-PML). Blood 90 (10): 4046-53, 1997.
- Gomis F, Sanz J, Sempere A, et al.: Immunofluorescent analysis with the anti-PML monoclonal antibody PG-M3 for rapid and accurate genetic diagnosis of acute promyelocytic leukemia. Ann Hematol 83 (11): 687-90, 2004.
- Dimov ND, Medeiros LJ, Kantarjian HM, et al.: Rapid and reliable confirmation of acute promyelocytic leukemia by immunofluorescence staining with an antipromyelocytic leukemia antibody: the M. D. Anderson Cancer Center experience of 349 patients. Cancer 116 (2): 369-76, 2010.
- Tallman MS, Hakimian D, Kwaan HC, et al.: New insights into the pathogenesis of coagulation dysfunction in acute promyelocytic leukemia. Leuk Lymphoma 11 (1-2): 27-36, 1993.
- Altman JK, Rademaker A, Cull E, et al.: Administration of ATRA to newly diagnosed patients with acute promyelocytic leukemia is delayed contributing to early hemorrhagic death. Leuk Res 37 (9): 1004-9, 2013.
- Lehmann S, Ravn A, Carlsson L, et al.: Continuing high early death rate in acute promyelocytic leukemia: a population-based report from the Swedish Adult Acute Leukemia Registry. Leukemia 25 (7): 1128-34, 2011.
- Park JH, Qiao B, Panageas KS, et al.: Early death rate in acute promyelocytic leukemia remains high despite all-trans retinoic acid. Blood 118 (5): 1248-54, 2011.
- Abla O, Ribeiro RC, Testi AM, et al.: Predictors of thrombohemorrhagic early death in children and adolescents with t(15;17)-positive acute promyelocytic leukemia treated with ATRA and chemotherapy. Ann Hematol 96 (9): 1449-1456, 2017.
- Breen KA, Grimwade D, Hunt BJ: The pathogenesis and management of the coagulopathy of acute promyelocytic leukaemia. Br J Haematol 156 (1): 24-36, 2012.
- Visani G, Gugliotta L, Tosi P, et al.: All-trans retinoic acid significantly reduces the incidence of early hemorrhagic death during induction therapy of acute promyelocytic leukemia. Eur J Haematol 64 (3): 139-44, 2000.
- de Botton S, Coiteux V, Chevret S, et al.: Outcome of childhood acute promyelocytic leukemia with all-trans-retinoic acid and chemotherapy. J Clin Oncol 22 (8): 1404-12, 2004.
- Testi AM, Biondi A, Lo Coco F, et al.: GIMEMA-AIEOPAIDA protocol for the treatment of newly diagnosed acute promyelocytic leukemia (APL) in children. Blood 106 (2): 447-53, 2005.
- Ortega JJ, Madero L, Martín G, et al.: Treatment with all-trans retinoic acid and anthracycline monochemotherapy for children with acute promyelocytic leukemia: a multicenter study by the PETHEMA Group. J Clin Oncol 23 (30): 7632-40, 2005.
- Guglielmi C, Martelli MP, Diverio D, et al.: Immunophenotype of adult and childhood acute promyelocytic leukaemia: correlation with morphology, type of PML gene breakpoint and clinical outcome. A cooperative Italian study on 196 cases. Br J Haematol 102 (4): 1035-41, 1998.
- Sanz MA, Lo Coco F, Martín G, et al.: Definition of relapse risk and role of nonanthracycline drugs for consolidation in patients with acute promyelocytic leukemia: a joint study of the PETHEMA and GIMEMA cooperative groups. Blood 96 (4): 1247-53, 2000.
- Sanz MA, Martín G, González M, et al.: Risk-adapted treatment of acute promyelocytic leukemia with all-trans-retinoic acid and anthracycline monochemotherapy: a multicenter study by the PETHEMA group. Blood 103 (4): 1237-43, 2004.
- Lo-Coco F, Avvisati G, Vignetti M, et al.: Front-line treatment of acute promyelocytic leukemia with AIDA induction followed by risk-adapted consolidation for adults younger than 61 years: results of the AIDA-2000 trial of the GIMEMA Group. Blood 116 (17): 3171-9, 2010.
- Callens C, Chevret S, Cayuela JM, et al.: Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia 19 (7): 1153-60, 2005.
- Gale RE, Hills R, Pizzey AR, et al.: Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood 106 (12): 3768-76, 2005.
- Arrigoni P, Beretta C, Silvestri D, et al.: FLT3 internal tandem duplication in childhood acute myeloid leukaemia: association with hyperleucocytosis in acute promyelocytic leukaemia. Br J Haematol 120 (1): 89-92, 2003.
- Noguera NI, Breccia M, Divona M, et al.: Alterations of the FLT3 gene in acute promyelocytic leukemia: association with diagnostic characteristics and analysis of clinical outcome in patients treated with the Italian AIDA protocol. Leukemia 16 (11): 2185-9, 2002.
- Tallman MS, Kim HT, Montesinos P, et al.: Does microgranular variant morphology of acute promyelocytic leukemia independently predict a less favorable outcome compared with classical M3 APL? A joint study of the North American Intergroup and the PETHEMA Group. Blood 116 (25): 5650-9, 2010.
- Iland HJ, Bradstock K, Supple SG, et al.: All-trans-retinoic acid, idarubicin, and IV arsenic trioxide as initial therapy in acute promyelocytic leukemia (APML4). Blood 120 (8): 1570-80; quiz 1752, 2012.
- Kutny MA, Moser BK, Laumann K, et al.: FLT3 mutation status is a predictor of early death in pediatric acute promyelocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 59 (4): 662-7, 2012.
- Kutny MA, Alonzo TA, Gerbing RB, et al.: Arsenic Trioxide Consolidation Allows Anthracycline Dose Reduction for Pediatric Patients With Acute Promyelocytic Leukemia: Report From the Children's Oncology Group Phase III Historically Controlled Trial AAML0631. J Clin Oncol 35 (26): 3021-3029, 2017.
- Kutny MA, Alonzo TA, Abla O, et al.: Assessment of Arsenic Trioxide and All-trans Retinoic Acid for the Treatment of Pediatric Acute Promyelocytic Leukemia: A Report From the Children's Oncology Group AAML1331 Trial. JAMA Oncol 8 (1): 79-87, 2022.
- de Botton S, Sanz MA, Chevret S, et al.: Extramedullary relapse in acute promyelocytic leukemia treated with all-trans retinoic acid and chemotherapy. Leukemia 20 (1): 35-41, 2006.
- Montesinos P, Díaz-Mediavilla J, Debén G, et al.: Central nervous system involvement at first relapse in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline monochemotherapy without intrathecal prophylaxis. Haematologica 94 (9): 1242-9, 2009.
- Chow J, Feusner J: Isolated central nervous system recurrence of acute promyelocytic leukemia in children. Pediatr Blood Cancer 52 (1): 11-3, 2009.
- Kaspers G, Gibson B, Grimwade D, et al.: Central nervous system involvement in relapsed acute promyelocytic leukemia. Pediatr Blood Cancer 53 (2): 235-6; author reply 237, 2009.
- Lo-Coco F, Avvisati G, Vignetti M, et al.: Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 369 (2): 111-21, 2013.
- Platzbecker U, Avvisati G, Cicconi L, et al.: Improved Outcomes With Retinoic Acid and Arsenic Trioxide Compared With Retinoic Acid and Chemotherapy in Non-High-Risk Acute Promyelocytic Leukemia: Final Results of the Randomized Italian-German APL0406 Trial. J Clin Oncol 35 (6): 605-612, 2017.
- Creutzig U, Dworzak MN, Bochennek K, et al.: First experience of the AML-Berlin-Frankfurt-Münster group in pediatric patients with standard-risk acute promyelocytic leukemia treated with arsenic trioxide and all-trans retinoid acid. Pediatr Blood Cancer 64 (8): , 2017.
- Yang MH, Wan WQ, Luo JS, et al.: Multicenter randomized trial of arsenic trioxide and Realgar-Indigo naturalis formula in pediatric patients with acute promyelocytic leukemia: Interim results of the SCCLG-APL clinical study. Am J Hematol 93 (12): 1467-1473, 2018.
- Zhang L, Zou Y, Chen Y, et al.: Role of cytarabine in paediatric acute promyelocytic leukemia treated with the combination of all-trans retinoic acid and arsenic trioxide: a randomized controlled trial. BMC Cancer 18 (1): 374, 2018.
- Zheng H, Jiang H, Hu S, et al.: Arsenic Combined With All-Trans Retinoic Acid for Pediatric Acute Promyelocytic Leukemia: Report From the CCLG-APL2016 Protocol Study. J Clin Oncol 39 (28): 3161-3170, 2021.
- Altucci L, Rossin A, Raffelsberger W, et al.: Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat Med 7 (6): 680-6, 2001.
- Huang ME, Ye YC, Chen SR, et al.: Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 72 (2): 567-72, 1988.
- Castaigne S, Chomienne C, Daniel MT, et al.: All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood 76 (9): 1704-9, 1990.
- Imaizumi M, Tawa A, Hanada R, et al.: Prospective study of a therapeutic regimen with all-trans retinoic acid and anthracyclines in combination of cytarabine in children with acute promyelocytic leukaemia: the Japanese childhood acute myeloid leukaemia cooperative study. Br J Haematol 152 (1): 89-98, 2011.
- Gregory J, Kim H, Alonzo T, et al.: Treatment of children with acute promyelocytic leukemia: results of the first North American Intergroup trial INT0129. Pediatr Blood Cancer 53 (6): 1005-10, 2009.
- Testi AM, Pession A, Diverio D, et al.: Risk-adapted treatment of acute promyelocytic leukemia: results from the International Consortium for Childhood APL. Blood 132 (4): 405-412, 2018.
- Sanz MA, Montesinos P, Rayón C, et al.: Risk-adapted treatment of acute promyelocytic leukemia based on all-trans retinoic acid and anthracycline with addition of cytarabine in consolidation therapy for high-risk patients: further improvements in treatment outcome. Blood 115 (25): 5137-46, 2010.
- Powell BL, Moser B, Stock W, et al.: Arsenic trioxide improves event-free and overall survival for adults with acute promyelocytic leukemia: North American Leukemia Intergroup Study C9710. Blood 116 (19): 3751-7, 2010.
- Shen ZX, Shi ZZ, Fang J, et al.: All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A 101 (15): 5328-35, 2004.
- Ravandi F, Estey E, Jones D, et al.: Effective treatment of acute promyelocytic leukemia with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab ozogamicin. J Clin Oncol 27 (4): 504-10, 2009.
- Hu J, Liu YF, Wu CF, et al.: Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A 106 (9): 3342-7, 2009.
- Cheng Y, Zhang L, Wu J, et al.: Long-term prognosis of childhood acute promyelocytic leukaemia with arsenic trioxide administration in induction and consolidation chemotherapy phases: a single-centre experience. Eur J Haematol 91 (6): 483-9, 2013.
- Wang H, Chen XY, Wang BS, et al.: The efficacy and safety of arsenic trioxide with or without all-trans retinoic acid for the treatment of acute promyelocytic leukemia: a meta-analysis. Leuk Res 35 (9): 1170-7, 2011.
- Zhang L, Zhao H, Zhu X, et al.: Retrospective analysis of 65 Chinese children with acute promyelocytic leukemia: a single center experience. Pediatr Blood Cancer 51 (2): 210-5, 2008.
- Zhou J, Zhang Y, Li J, et al.: Single-agent arsenic trioxide in the treatment of children with newly diagnosed acute promyelocytic leukemia. Blood 115 (9): 1697-702, 2010.
- Iland HJ, Collins M, Bradstock K, et al.: Use of arsenic trioxide in remission induction and consolidation therapy for acute promyelocytic leukaemia in the Australasian Leukaemia and Lymphoma Group (ALLG) APML4 study: a non-randomised phase 2 trial. Lancet Haematol 2 (9): e357-66, 2015.
- Douer D, Zickl LN, Schiffer CA, et al.: All-trans retinoic acid and late relapses in acute promyelocytic leukemia: very long-term follow-up of the North American Intergroup Study I0129. Leuk Res 37 (7): 795-801, 2013.
- Coombs CC, DeAngelis LM, Feusner JH, et al.: Pseudotumor Cerebri in Acute Promyelocytic Leukemia Patients on Intergroup Protocol 0129: Clinical Description and Recommendations for New Diagnostic Criteria. Clin Lymphoma Myeloma Leuk 16 (3): 146-51, 2016.
- Sanz MA, Montesinos P: How we prevent and treat differentiation syndrome in patients with acute promyelocytic leukemia. Blood 123 (18): 2777-82, 2014.
- Montesinos P, Bergua JM, Vellenga E, et al.: Differentiation syndrome in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline chemotherapy: characteristics, outcome, and prognostic factors. Blood 113 (4): 775-83, 2009.
- Mitrovic M, Suvajdzic N, Bogdanovic A, et al.: International Society of Thrombosis and Hemostasis Scoring System for disseminated intravascular coagulation ≥ 6: a new predictor of hemorrhagic early death in acute promyelocytic leukemia. Med Oncol 30 (1): 478, 2013.
- Rajpurkar M, Alonzo TA, Wang YC, et al.: Risk Markers for Significant Bleeding and Thrombosis in Pediatric Acute Promyelocytic Leukemia; Report From the Children's Oncology Group Study AAML0631. J Pediatr Hematol Oncol 41 (1): 51-55, 2019.
- Unnikrishnan D, Dutcher JP, Varshneya N, et al.: Torsades de pointes in 3 patients with leukemia treated with arsenic trioxide. Blood 97 (5): 1514-6, 2001.
- Barbey JT: Cardiac toxicity of arsenic trioxide. Blood 98 (5): 1632; discussion 1633-4, 2001.
- Jurcic JG, Nimer SD, Scheinberg DA, et al.: Prognostic significance of minimal residual disease detection and PML/RAR-alpha isoform type: long-term follow-up in acute promyelocytic leukemia. Blood 98 (9): 2651-6, 2001.
- Diverio D, Rossi V, Avvisati G, et al.: Early detection of relapse by prospective reverse transcriptase-polymerase chain reaction analysis of the PML/RARalpha fusion gene in patients with acute promyelocytic leukemia enrolled in the GIMEMA-AIEOP multicenter "AIDA" trial. GIMEMA-AIEOP Multicenter "AIDA" Trial. Blood 92 (3): 784-9, 1998.
- Lo Coco F, Diverio D, Avvisati G, et al.: Therapy of molecular relapse in acute promyelocytic leukemia. Blood 94 (7): 2225-9, 1999.
- Esteve J, Escoda L, Martín G, et al.: Outcome of patients with acute promyelocytic leukemia failing to front-line treatment with all-trans retinoic acid and anthracycline-based chemotherapy (PETHEMA protocols LPA96 and LPA99): benefit of an early intervention. Leukemia 21 (3): 446-52, 2007.
- Zelent A, Guidez F, Melnick A, et al.: Translocations of the RARalpha gene in acute promyelocytic leukemia. Oncogene 20 (49): 7186-203, 2001.
- Yan W, Zhang G: Molecular Characteristics and Clinical Significance of 12 Fusion Genes in Acute Promyelocytic Leukemia: A Systematic Review. Acta Haematol 136 (1): 1-15, 2016.
- Rego EM, Ruggero D, Tribioli C, et al.: Leukemia with distinct phenotypes in transgenic mice expressing PML/RAR alpha, PLZF/RAR alpha or NPM/RAR alpha. Oncogene 25 (13): 1974-9, 2006.
- Licht JD, Chomienne C, Goy A, et al.: Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood 85 (4): 1083-94, 1995.
- Guidez F, Ivins S, Zhu J, et al.: Reduced retinoic acid-sensitivities of nuclear receptor corepressor binding to PML- and PLZF-RARalpha underlie molecular pathogenesis and treatment of acute promyelocytic leukemia. Blood 91 (8): 2634-42, 1998.
- Grimwade D, Biondi A, Mozziconacci MJ, et al.: Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Français de Cytogénétique Hématologique, Groupe de Français d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies". Blood 96 (4): 1297-308, 2000.
- Sukhai MA, Wu X, Xuan Y, et al.: Myeloid leukemia with promyelocytic features in transgenic mice expressing hCG-NuMA-RARalpha. Oncogene 23 (3): 665-78, 2004.
- Redner RL, Corey SJ, Rush EA: Differentiation of t(5;17) variant acute promyelocytic leukemic blasts by all-trans retinoic acid. Leukemia 11 (7): 1014-6, 1997.
- Wells RA, Catzavelos C, Kamel-Reid S: Fusion of retinoic acid receptor alpha to NuMA, the nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat Genet 17 (1): 109-13, 1997.
- Wells RA, Hummel JL, De Koven A, et al.: A new variant translocation in acute promyelocytic leukaemia: molecular characterization and clinical correlation. Leukemia 10 (4): 735-40, 1996.
- Marjerrison S, Antillon F, Bonilla M, et al.: Outcome of children treated for relapsed acute myeloid leukemia in Central America. Pediatr Blood Cancer 61 (7): 1222-6, 2014.
- Lengfelder E, Lo-Coco F, Ades L, et al.: Arsenic trioxide-based therapy of relapsed acute promyelocytic leukemia: registry results from the European LeukemiaNet. Leukemia 29 (5): 1084-91, 2015.
- Holter Chakrabarty JL, Rubinger M, Le-Rademacher J, et al.: Autologous is superior to allogeneic hematopoietic cell transplantation for acute promyelocytic leukemia in second complete remission. Biol Blood Marrow Transplant 20 (7): 1021-5, 2014.
- Fox E, Razzouk BI, Widemann BC, et al.: Phase 1 trial and pharmacokinetic study of arsenic trioxide in children and adolescents with refractory or relapsed acute leukemia, including acute promyelocytic leukemia or lymphoma. Blood 111 (2): 566-73, 2008.
- Niu C, Yan H, Yu T, et al.: Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow-up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood 94 (10): 3315-24, 1999.
- Shen ZX, Chen GQ, Ni JH, et al.: Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 89 (9): 3354-60, 1997.
- Avvisati G, Lo-Coco F, Paoloni FP, et al.: AIDA 0493 protocol for newly diagnosed acute promyelocytic leukemia: very long-term results and role of maintenance. Blood 117 (18): 4716-25, 2011.
- Fouzia NA, Sharma V, Ganesan S, et al.: Management of relapse in acute promyelocytic leukaemia treated with up-front arsenic trioxide-based regimens. Br J Haematol 192 (2): 292-299, 2021.
- Lu J, Huang X, Bao L, et al.: Treatment outcomes in relapsed acute promyelocytic leukemia patients initially treated with all-trans retinoic acid and arsenic compound-based combined therapies. Oncol Lett 7 (1): 177-182, 2014.
- Zhu HH, Qin YZ, Huang XJ: Resistance to arsenic therapy in acute promyelocytic leukemia. N Engl J Med 370 (19): 1864-6, 2014.
- Soignet SL, Maslak P, Wang ZG, et al.: Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med 339 (19): 1341-8, 1998.
- Zhang P: The use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia. J Biol Regul Homeost Agents 13 (4): 195-200, 1999 Oct-Dec.
- Lo-Coco F, Cimino G, Breccia M, et al.: Gemtuzumab ozogamicin (Mylotarg) as a single agent for molecularly relapsed acute promyelocytic leukemia. Blood 104 (7): 1995-9, 2004.
- Dvorak CC, Agarwal R, Dahl GV, et al.: Hematopoietic stem cell transplant for pediatric acute promyelocytic leukemia. Biol Blood Marrow Transplant 14 (7): 824-30, 2008.
- Bourquin JP, Thornley I, Neuberg D, et al.: Favorable outcome of allogeneic hematopoietic stem cell transplantation for relapsed or refractory acute promyelocytic leukemia in childhood. Bone Marrow Transplant 34 (9): 795-8, 2004.
- de Botton S, Fawaz A, Chevret S, et al.: Autologous and allogeneic stem-cell transplantation as salvage treatment of acute promyelocytic leukemia initially treated with all-trans-retinoic acid: a retrospective analysis of the European acute promyelocytic leukemia group. J Clin Oncol 23 (1): 120-6, 2005.
- Meloni G, Diverio D, Vignetti M, et al.: Autologous bone marrow transplantation for acute promyelocytic leukemia in second remission: prognostic relevance of pretransplant minimal residual disease assessment by reverse-transcription polymerase chain reaction of the PML/RAR alpha fusion gene. Blood 90 (3): 1321-5, 1997.
- Thirugnanam R, George B, Chendamarai E, et al.: Comparison of clinical outcomes of patients with relapsed acute promyelocytic leukemia induced with arsenic trioxide and consolidated with either an autologous stem cell transplant or an arsenic trioxide-based regimen. Biol Blood Marrow Transplant 15 (11): 1479-84, 2009.
- Abla O, Kutny MA, Testi AM, et al.: Management of relapsed and refractory childhood acute promyelocytic leukaemia: recommendations from an international expert panel. Br J Haematol 175 (4): 588-601, 2016.
Myeloid Proliferations Associated With Down Syndrome
Myeloid leukemias that arise in children with Down syndrome, particularly in patients younger than 4 years, are a distinct subset of AML characterized by the co-existence of trisomy 21 and GATA1 mutations within the leukemic blasts that are often, but not always, megakaryoblastic. This distinct leukemia is further subdivided into two versions: a transient newborn and young-infant version called transient abnormal myelopoiesis (TAM), which spontaneously remits over time; and an unremitting but chemosensitive version that appears later, between the ages of 90 days and 3 years.[1] It is important to recognize the possibility of these versions in both children with Down syndrome phenotypes and in those who have mosaic trisomy 21, which can be solely present in the leukemic blasts. If possible, newborns with apparent AML should not have therapy initiated until genetic testing results have been returned.[2] In older children with megakaryocytic AML, it is important to rule out the presence of co-existing trisomy 21 and GATA1 mutations; these children may be successfully treated on the lower-intensity chemotherapy regimens that are used for children with myeloid leukemia associated with Down syndrome.[3]
Transient Abnormal Myelopoiesis (TAM) Associated With Down Syndrome
Approximately 10% of neonates with Down syndrome develop a TAM (also termed transient myeloproliferative disorder [TMD]).[2] This disorder mimics congenital AML but typically improves spontaneously within the first 3 months of life (median, 49 days), although TAM has been reported to remit as late as 20 months.[4] The late remissions likely reflect a persistent hepatomegaly from TAM-associated hepatic fibrosis rather than active disease.[5]
Although TAM is usually a self-resolving condition, it can be associated with significant morbidity and may be fatal in 10% to 17% of affected infants.[4,5,6,7,8] Infants with progressive organomegaly, visceral effusions, preterm delivery (less than 37 weeks of gestation), bleeding diatheses, failure of spontaneous remission, laboratory evidence of progressive liver dysfunction (elevated direct bilirubin), renal failure, and very high white blood cell (WBC) count are at particularly high risk of early mortality.[5,6,8] Death has been reported to occur in 21% of these patients with high-risk TAM, although only 10% were attributable to TAM and the remaining deaths were caused by coexisting conditions known to be more prominent in neonates with Down syndrome.[5]
The following three risk groups have been identified on the basis of the diagnostic clinical findings of hepatomegaly with or without life-threatening symptoms:[5]
- Low risk includes those with neither hepatomegaly nor life-threatening symptoms (38% of patients and an overall survival [OS] rate of 92% ± 8%).
- Intermediate risk includes those with hepatomegaly alone (40% of patients and an OS rate of 77% ± 12%).
- High risk includes those with hepatomegaly and life-threatening symptoms (21% of patients and an OS rate of 51% ± 19%).
Therapeutic intervention is warranted in patients with apparent severe hydrops or organ failure. Because TAM eventually spontaneously remits, treatment is short in duration and primarily aimed at the reduction of leukemic burden and resolution of immediate symptoms. Several treatment approaches have been used, including the following:
- Exchange transfusion.
- Leukapheresis.
- Low-dose cytarabine. Of these approaches, only cytarabine has been shown to consistently reduce TAM complications and related mortality.[5,8]; [9][Level of evidence B4] Cytarabine dosing has ranged from 0.4 to 1.5 mg/kg per dose given intravenously (IV) or subcutaneously (SC) once to twice daily for 4 to 12 days,[8] with similar efficacies and less toxicity than higher, continuous 5-day infusions, which led to prolonged severe neutropenia.[5] A prospective trial that utilized cytarabine at 1.5 mg/kg per day IV or SC for 7 days for symptomatic patients reported a significant reduction in early death compared with similar historical controls (12% ± 5% vs. 33% ± 7%, respectively; P = .2).[9][Level of evidence B4]
Subsequent development of myeloid leukemia associated with Down syndrome is seen in 10% to 30% of children who have a spontaneous remission of TAM and has been reported at a mean age of 16 months (range, 1–30 months).[4,5,10] While TAM is generally not characterized by cytogenetic abnormalities other than trisomy 21, the presence of additional cytogenetic findings may connote an increased risk of developing subsequent myeloid leukemia associated with Down syndrome.[6] An additional risk factor reported in two studies is the late resolution of TAM, measured by either time to complete resolution of signs of TAM (defined as resolution beyond the median, 47 days from diagnosis) or by persistence of minimal residual disease (MRD) in the peripheral blood at week 12 of follow-up.[5]; [9][Level of evidence B4] The use of cytarabine for TAM symptoms or persistent MRD in TAM has failed to show a reduction in later myeloid leukemia associated with Down syndrome, as reported in large observational cohort studies.[5,8] In a prospective single-arm trial designed to assess whether cytarabine treatment for TAM could prevent the development of later myeloid leukemia associated with Down syndrome, no benefit was found when compared with historical controls (19% ± 4% vs. 22% ± 4%, respectively; P = .88).[9][Level of evidence B4]
Myeloid Leukemia Associated With Down Syndrome
Children with Down syndrome have a 10-fold to 45-fold increased risk of leukemia when compared with children without Down syndrome.[11] However, the ratio of acute lymphoblastic leukemia to acute myeloid leukemia (AML) is typical for childhood acute leukemia. The exception is during the first 3 years of life, when AML, particularly the megakaryoblastic subtype, predominates and exhibits a distinctive biology characterized by GATA1 mutations and increased sensitivity to cytarabine.[12,13,14,15,16,17] Importantly, these risks appear to be similar whether a child has phenotypic characteristics of Down syndrome or whether a child has only genetic bone marrow mosaicism.[18]
Prognosis and Treatment of Children With Down Syndrome and AML
Outcome is generally favorable for children with Down syndrome who develop AML (called myeloid leukemia associated with Down syndrome in the World Health Organization classification).[19,20,21]
Prognostic factors for children with Down syndrome and AML include the following:
- Age. The prognosis is particularly good (EFS rates exceeding 85%) in children aged 4 years or younger at diagnosis; this age group accounts for the vast majority of Down syndrome patients with AML.[20,21,22,23] Children with Down syndrome who are older than 4 years have a significantly worse prognosis and warrant therapy utilized in children with AML without Down syndrome unless a GATA1 mutation is found.[24]
- White blood cell count. A large international Berlin-Frankfurt-Münster (BFM) retrospective study of 451 children with AML and Down syndrome (aged >6 months and <5 years) observed a 7-year EFS rate of 78% and a 7-year OS rate of 79%. In multivariate analyses, WBC count (≥20 × 109 /L) and age (>3 years) were independent predictors of lower EFS. The 7-year EFS rate for the older population (>3 years) and for the higher WBC-count population still exceeded 60%.[25]
- AML karyotype. Normal karyotypic AML (other than trisomy 21), which was observed in 29% of patients, independently predicted for inferior OS and EFS (7-year EFS rate of 65% compared with 82% for patients with aberrant karyotypes).[25] However, this was not seen in a later trial.[23] In this same trial, the presence of trisomy 8 was shown to adversely impact prognosis. In another study, complex karyotypes (≥3 independent abnormalities) were prognostic of an increased cumulative incidence of relapse at 2 years (30.8% compared with 7.5% in patients without complex karyotypes; P = .001).[26]
- MRD. MRD at the end of induction 1 was found to be a strong prognostic factor;[21,27] this was consistent with the BFM finding that early response correlated with improved OS.[23] However, negative MRD at the end of induction 1 did not identify a group of patients who could receive reduced chemotherapy.[26]
Approximately 29% to 47% of Down syndrome patients present with myelodysplastic syndromes (MDS) (<20% blasts) but their outcomes are similar to those with AML.[20,21,23]
Treatment options for newly diagnosed children with Down syndrome and AML include the following:
- Chemotherapy.
Appropriate therapy for younger children (aged ≤4 years) with Down syndrome and AML is less intensive than current standard childhood AML therapy. Hematopoietic stem cell transplant is not indicated in first remission.[10,14,19,20,21,22,23,24,28]
Evidence (chemotherapy):
- In a Children's Oncology Group (COG) trial for newly diagnosed children with Down syndrome and AML (AAML0431 [NCT00369317]), 204 children were enrolled on a regimen that substituted high-dose cytarabine for the second of four induction cycles (thereby reducing cumulative anthracycline exposure from 320 mg to 240 mg), moving this cycle from intensification where it was used in the previous COG A2971 (NCT00003593) trial.[20,21] Intrathecal doses were reduced from seven to two total injections and intensification included two cycles of cytarabine/etoposide.
- When compared with the previous trial, these changes resulted in an overall improvement of approximately 10%.
- The EFS rate was 89.9%, and the OS rate was 93%.
- Relapse occurred in 14 patients and there were two treatment-related deaths, both related to pneumonia, neither of which occurred during induction 2.
- No patient had central nervous system (CNS) involvement on this trial or the preceding COG A2971 (NCT00003593) trial.[20]
- The only prognostic factor identified was MRD using flow cytometry on day 28 of induction 1. Among those who were MRD negative (≤0.01%), the DFS rate was 92.7%; in the 14.4% of patients who were MRD positive, the DFS rate was 76.2% (P = .011).
- In a joint trial (ML-DS 2006) from the BFM, Dutch Childhood Oncology Group (DCOG), and Nordic Society of Pediatric Hematology and Oncology (NOPHO), 170 children with Down syndrome were enrolled in a trial that focused on reducing therapy by eliminating etoposide during consolidation, reducing the number of intrathecal doses from 11 to 4, and the elimination of maintenance from the reduced therapy Down syndrome arm of AML-BFM 98.[23] As in the COG trials, no patient had CNS disease at diagnosis.
- Outcomes were no worse despite reduction in chemotherapy. The OS rate was 89% (± 3%) and the EFS rate was 87% (± 3%), similar to that observed in AML-BFM 98 (OS rate, 90% ± 4% [P = NS]; EFS rate, 89% ± 4% [P = NS]). The cumulative incidence of relapse (CIR) rate was 6% in both trials.
- Nine patients relapsed, and seven of those patients died.
- Patients with a good early response (<5% blasts by morphology before induction cycle 2, n = 123 [72%]) had better outcomes (OS rate, 92% ± 3% vs. 57% ± 16%, P < .0001; EFS rate, 88% ± 3% vs. 58% ± 16%, P = .0008; and CIR rate, 3% ± 2% vs. 27% ± 18%, P = .003).
- Less toxicity was seen in this new trial, and treatment-related mortality remained low (2.9% vs. 5%, P = .276).
The following two prognostic factors were identified:[23]
- Trisomy 8 was an adverse factor (n = 37; OS rate, 77% vs. 95%, P = .07; EFS rate, 73% ± 8% vs. 91% ± 4%, P = .018; CIR rate, 16% ± 7% vs. 3% ± 2%, P = .02).
- This was confirmed in multivariate analysis, where lack of good early response and trisomy 8 maintained their adverse impact on relapse, with relative risks of 8.55 (95% confidence interval [CI], 1.96–37.29, P = .004) and 4.36 (1.24–15.39, P = .022), respectively.
Children with mosaicism for trisomy 21 are treated similarly to those children with clinically evident Down syndrome.[5,18,20] Although an optimal treatment for these children has not been defined, they are usually treated on AML regimens designed for children without Down syndrome.
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Refractory Disease or Relapse in Children With Down Syndrome
A small number of publications address outcomes in children with Down syndrome who relapse after initial therapy or who have refractory AML. In three prospective trials of children with Down Syndrome and newly diagnosed AML, outcomes were poor for those who relapsed (4 of 11, 2 of 9, and 2 of 12 patients who relapsed survived).[19,23,26] Thus, these children are treated similarly to children without Down syndrome, with an intensive reinduction chemotherapy regimen, and if a remission is achieved, therapy is followed by an allogeneic hematopoietic stem cell transplantation (HSCT).
Treatment options for children with Down syndrome with refractory or relapsed AML include the following:
- Chemotherapy, which may be followed by an allogeneic HSCT.
Evidence (treatment of children with Down syndrome with refractory or relapsed AML):
- The Japanese Pediatric Leukemia/Lymphoma Study Group reported the outcomes of 29 patients with Down syndrome and relapsed (n = 26) or refractory (n = 3) AML. As expected with Down syndrome, the children in this cohort were very young (median age, 2 years); relapses were almost all early (median, 8.6 months; 80% <12 months from diagnosis); and 89% had M7 French-American-British classification.[29][Level of evidence C1]
- In contrast to the excellent outcomes achieved after initial therapy, only 50% of the children attained a second remission, and the 3-year OS rate was 26%.
- Approximately one-half of the children underwent allogeneic transplant, and no advantage was noted with transplant compared with chemotherapy, but the number of patients was small.
- A Center for International Blood and Marrow Transplant Research study of children with Down syndrome and AML who underwent HSCT reported the following results:[30][Level of evidence C1]
- A similarly poor outcome, with a 3-year OS rate of 19%.
- The main cause of failure after transplant was relapse, which exceeded 60%; transplant-related mortality was approximately 20%.
- A Japanese registry study reported better survival after transplant of children with Down Syndrome using reduced-intensity conditioning regimens compared with myeloablative approaches, but the number of patients was very small (n = 5) and the efficacy of reduced-intensity approaches in children with Down syndrome and AML requires further study.[31][Level of evidence C2]
References:
- Lange B: The management of neoplastic disorders of haematopoiesis in children with Down's syndrome. Br J Haematol 110 (3): 512-24, 2000.
- Gamis AS, Smith FO: Transient myeloproliferative disorder in children with Down syndrome: clarity to this enigmatic disorder. Br J Haematol 159 (3): 277-87, 2012.
- de Rooij JD, Branstetter C, Ma J, et al.: Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet 49 (3): 451-456, 2017.
- Homans AC, Verissimo AM, Vlacha V: Transient abnormal myelopoiesis of infancy associated with trisomy 21. Am J Pediatr Hematol Oncol 15 (4): 392-9, 1993.
- Gamis AS, Alonzo TA, Gerbing RB, et al.: Natural history of transient myeloproliferative disorder clinically diagnosed in Down syndrome neonates: a report from the Children's Oncology Group Study A2971. Blood 118 (26): 6752-9; quiz 6996, 2011.
- Massey GV, Zipursky A, Chang MN, et al.: A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood 107 (12): 4606-13, 2006.
- Muramatsu H, Kato K, Watanabe N, et al.: Risk factors for early death in neonates with Down syndrome and transient leukaemia. Br J Haematol 142 (4): 610-5, 2008.
- Klusmann JH, Creutzig U, Zimmermann M, et al.: Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood 111 (6): 2991-8, 2008.
- Flasinski M, Scheibke K, Zimmermann M, et al.: Low-dose cytarabine to prevent myeloid leukemia in children with Down syndrome: TMD Prevention 2007 study. Blood Adv 2 (13): 1532-1540, 2018.
- Ravindranath Y, Abella E, Krischer JP, et al.: Acute myeloid leukemia (AML) in Down's syndrome is highly responsive to chemotherapy: experience on Pediatric Oncology Group AML Study 8498. Blood 80 (9): 2210-4, 1992.
- Marlow EC, Ducore J, Kwan ML, et al.: Leukemia Risk in a Cohort of 3.9 Million Children with and without Down Syndrome. J Pediatr 234: 172-180.e3, 2021.
- Ravindranath Y: Down syndrome and leukemia: new insights into the epidemiology, pathogenesis, and treatment. Pediatr Blood Cancer 44 (1): 1-7, 2005.
- Ross JA, Spector LG, Robison LL, et al.: Epidemiology of leukemia in children with Down syndrome. Pediatr Blood Cancer 44 (1): 8-12, 2005.
- Gamis AS: Acute myeloid leukemia and Down syndrome evolution of modern therapy--state of the art review. Pediatr Blood Cancer 44 (1): 13-20, 2005.
- Taub JW, Ge Y: Down syndrome, drug metabolism and chromosome 21. Pediatr Blood Cancer 44 (1): 33-9, 2005.
- Crispino JD: GATA1 mutations in Down syndrome: implications for biology and diagnosis of children with transient myeloproliferative disorder and acute megakaryoblastic leukemia. Pediatr Blood Cancer 44 (1): 40-4, 2005.
- Ge Y, Stout ML, Tatman DA, et al.: GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst 97 (3): 226-31, 2005.
- Kudo K, Hama A, Kojima S, et al.: Mosaic Down syndrome-associated acute myeloid leukemia does not require high-dose cytarabine treatment for induction and consolidation therapy. Int J Hematol 91 (4): 630-5, 2010.
- Lange BJ, Kobrinsky N, Barnard DR, et al.: Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: Children's Cancer Group Studies 2861 and 2891. Blood 91 (2): 608-15, 1998.
- Sorrell AD, Alonzo TA, Hilden JM, et al.: Favorable survival maintained in children who have myeloid leukemia associated with Down syndrome using reduced-dose chemotherapy on Children's Oncology Group trial A2971: a report from the Children's Oncology Group. Cancer 118 (19): 4806-14, 2012.
- Taub JW, Berman JN, Hitzler JK, et al.: Improved outcomes for myeloid leukemia of Down syndrome: a report from the Children's Oncology Group AAML0431 trial. Blood 129 (25): 3304-3313, 2017.
- Creutzig U, Reinhardt D, Diekamp S, et al.: AML patients with Down syndrome have a high cure rate with AML-BFM therapy with reduced dose intensity. Leukemia 19 (8): 1355-60, 2005.
- Uffmann M, Rasche M, Zimmermann M, et al.: Therapy reduction in patients with Down syndrome and myeloid leukemia: the international ML-DS 2006 trial. Blood 129 (25): 3314-3321, 2017.
- Gamis AS, Woods WG, Alonzo TA, et al.: Increased age at diagnosis has a significantly negative effect on outcome in children with Down syndrome and acute myeloid leukemia: a report from the Children's Cancer Group Study 2891. J Clin Oncol 21 (18): 3415-22, 2003.
- Blink M, Zimmermann M, von Neuhoff C, et al.: Normal karyotype is a poor prognostic factor in myeloid leukemia of Down syndrome: a retrospective, international study. Haematologica 99 (2): 299-307, 2014.
- Hitzler J, Alonzo T, Gerbing R, et al.: High-dose AraC is essential for the treatment of ML-DS independent of postinduction MRD: results of the COG AAML1531 trial. Blood 138 (23): 2337-2346, 2021.
- Taga T, Tanaka S, Hasegawa D, et al.: Post-induction MRD by FCM and GATA1-PCR are significant prognostic factors for myeloid leukemia of Down syndrome. Leukemia 35 (9): 2508-2516, 2021.
- Taga T, Shimomura Y, Horikoshi Y, et al.: Continuous and high-dose cytarabine combined chemotherapy in children with down syndrome and acute myeloid leukemia: Report from the Japanese children's cancer and leukemia study group (JCCLSG) AML 9805 down study. Pediatr Blood Cancer 57 (1): 36-40, 2011.
- Taga T, Saito AM, Kudo K, et al.: Clinical characteristics and outcome of refractory/relapsed myeloid leukemia in children with Down syndrome. Blood 120 (9): 1810-5, 2012.
- Hitzler JK, He W, Doyle J, et al.: Outcome of transplantation for acute myelogenous leukemia in children with Down syndrome. Biol Blood Marrow Transplant 19 (6): 893-7, 2013.
- Muramatsu H, Sakaguchi H, Taga T, et al.: Reduced intensity conditioning in allogeneic stem cell transplantation for AML with Down syndrome. Pediatr Blood Cancer 61 (5): 925-7, 2014.
Myelodysplastic Syndromes (MDS)
Myelodysplastic syndromes (MDS) and myeloproliferative syndromes (MPS) represent between 5% and 10% of all myeloid malignancies in children. They are a heterogeneous group of disorders, with MDS usually presenting with cytopenias and MPS presenting with increased peripheral white blood cell, red blood cell, or platelet counts. MDS is characterized by ineffective hematopoiesis and increased cell death, while MPS is associated with increased progenitor proliferation and survival. Because they both represent disorders of very primitive, multipotential hematopoietic stem cells, curative therapeutic approaches nearly always require allogeneic hematopoietic stem cell transplantation (HSCT).
Risk Factors
Patients with the following germline mutations or inherited disorders have a significantly increased risk of developing MDS:
- Fanconi anemia: Caused by germline mutations in DNA repair genes.
- Dyskeratosis congenita: Resulting from mutations in genes regulating telomere length. Genes mutated in dyskeratosis congenita include ACD, CTC1, DKC1, NHP2, NOP10, PARN, RTEL1, TERC, TERT, TINF2, and WRAP53.
- Shwachman-Diamond syndrome, Diamond-Blackfan anemia, and other bone marrow failure syndromes: Resulting from mutations in genes encoding ribosome-associated proteins.[1,2]GATA1 mutations have been linked to Diamond-Blackfan anemia and MDS predisposition.[3]
- Severe congenital neutropenia: Caused by mutations in the gene encoding elastase. The 15-year cumulative risk of MDS in patients with severe congenital neutropenia, also known as Kostmann syndrome, has been estimated to be 15%, with an annual risk of MDS/acute myeloid leukemia (AML) of 2% to 3%. It is unclear how mutations affecting this protein and how the chronic exposure of granulocyte colony-stimulating factor (G-CSF) contribute to the development of MDS.[4]
- Trisomy 21 syndrome: GATA1 mutations are nearly always present in the transient leukemia associated with Trisomy 21 and MDS in children younger than 3 years with Down syndrome.[5]
- Congenital amegakaryocytic thrombocytopenia (CAMT): Inherited mutations in the RUNX1 or CEPBA genes are associated with CAMT.[6,7] Mutations in the MPL gene are the underlying genetic cause of CAMT; there is a less than 10% risk of developing MDS/AML in patients with CAMT.[8]
- GATA2 mutations: Germline mutations of GATA2 have been reported in patients with MDS/AML in conjunction with monocytopenia, B cell and natural killer cell deficiency, pulmonary alveolar proteinosis, and susceptibility to opportunistic infections.[9,10]
- RUNX1 or CEPBA mutations: Inherited mutations in the RUNX1 or CEPBA genes are associated with familial MDS/AML.[6,7]
- SAMD9 and SAMD9L mutations: Inherited mutations in SAMD9 and SAMD9L are associated with familial MDS.[11,12,13,14,15,16]
A retrospective analysis that used a capture assay to target mutations known to predispose to marrow failure and MDS was performed on genomic DNA from peripheral blood mononuclear cell samples from patients undergoing HSCT for MDS and aplastic anemia. Among the 46 children aged 18 years and younger with MDS, 10 patients (22%) harbored constitutional predisposition genetic mutations (5 GATA2, 1 each of MPL, RTEL1, SBDS, TINF2, and TP53), of which only 2 were suspected before transplant. This is considered a high incidence of genetic mutations compared with only 8% (4 of 64) in patients aged 18 to 40 years.[17]
Clinical Presentation
Patients usually present with signs of cytopenias, including pallor, infection, or bruising.
The bone marrow is usually characterized by hypercellularity and dysplastic changes in myeloid precursors. Clonal evolution can eventually lead to the development of AML. The percentage of abnormal blasts is less than 20% and lack common AML recurrent cytogenetic abnormalities (t(8;21), inv(16), t(15;17), or KMT2A translocations).
The less common hypocellular MDS can be distinguished from aplastic anemia in part by its marked dysplasia, clonal nature, and higher percentage of CD34-positive precursors.[18,19]
Molecular Abnormalities
Molecular features of myelodysplastic syndromes (MDS)
Pediatric MDS are associated with a distinctive constellation of genetic alterations compared with MDS arising in adults. In adults, MDS often evolves from clonal hematopoiesis and is characterized by mutations in TET2, DNMT3A, and TP53. In contrast, mutations in these genes are rare in pediatric MDS, while mutations in GATA2, SAMD9, SAMD9L, SETBP1, ASXL1, and RAS/MAPK pathway genes are observed in subsets of pediatric MDS cases.[11,20]
A report of the genomic landscape of pediatric MDS described the results of whole-exome sequencing for 32 pediatric patients with primary MDS and targeted sequencing for another 14 cases.[11] These 46 cases were equally divided between refractory cytopenia of childhood and MDS with excess blasts (MDS-EB). The results from the report include the following:
- Mutations in RAS/MAPK pathway genes were observed in 43% of primary MDS cases, with mutations most commonly involving PTPN11 and NRAS but with mutations also observed in other pathway members (e.g., BRAF [non–BRAF V600E], CBL, and KRAS). RAS/MAPK mutations were more common in patients with MDS-EB (65%) than in patients with refractory cytopenia of childhood (17%).
- Germline variants in SAMD9 (n = 4) or SAMD9L (n = 4) were observed in 17% of patients with primary MDS, with seven of eight mutations occurring in patients with refractory cytopenia of childhood. These cases all showed loss of material on chromosome 7. Approximately 40% of patients with deletions of part or all of chromosome 7 had germline SAMD9 or SAMD9L variants.
- GATA2 mutations were observed in three cases (7%), and all cases were confirmed or presumed to be germline.
- Deletions involving chromosome 7 were the most common copy number alteration and were observed in 41% of cases. Loss of part or all of chromosome 7 was most commonly observed in SAMD9 and SAMD9L cases (100%) and in MDS-EB patients with a RAS/MAPK mutation (71%).
- Other genes that were mutated in more than 1 of the 46 cases studied included SETBP1, ETV6, and TP53.
A second report described the application of a targeted sequencing panel of 105 genes to 50 pediatric patients with MDS (refractory cytopenia of childhood = 31 and MDS-EB = 19) and was enriched for cases with monosomy 7 (48%).[11,20]SAMD9 and SAMD9L were not included in the gene panel. The second report described the following results:
- Germline GATA2 mutations were observed in 30% of patients, and RUNX1 mutations were observed in 6% of patients.
- Somatic mutations were observed in 34% of patients and were more common in patients with MDS-EB than in patients with refractory cytopenia of childhood (68% vs. 13%).
- The most commonly mutated gene was SETBP1 (18%); less commonly mutated genes included ASXL1, RUNX1, and RAS/MAPK pathway genes (PTPN11, NRAS, KRAS, NF1). Twelve percent of cases showed mutations in RAS/MAPK pathway genes.
Patients with germline GATA2 mutations, in addition to MDS, show a wide range of hematopoietic and immune defects as well as nonhematopoietic manifestations.[21] The former defects include monocytopenia with susceptibility to atypical mycobacterial infection and DCML deficiency (loss of dendritic cells, monocytes, and B and natural killer lymphoid cells). The resulting immunodeficiency leads to increased susceptibility to warts, severe viral infections, mycobacterial infections, fungal infections, and human papillomavirus–related cancers. The nonhematopoietic manifestations include deafness and lymphedema.
Germline GATA2 mutations were studied in 426 pediatric patients with primary MDS and 82 cases with secondary MDS who were enrolled in consecutive studies of the European Working Group of MDS in Childhood (EWOG-MDS).[22] The study had the following results:
- Germline GATA2 mutations were identified in 7% of pediatric patients with primary MDS. While the median age of patients presenting with GATA2 mutations was 12.3 years in the EWOG-MDS pediatric population, most cases of germline GATA2-related myeloid neoplasms occur during adulthood.[23]
- GATA2 mutations were more common in patients with MDS-EB (15%) than in patients with refractory cytopenia of childhood (4%).
- Among patients with GATA2 mutations, 46% presented with MDS-EB and 70% showed monosomy 7.
- Familial MDS/AML was identified in 12 of 53 GATA2-mutated patients for whom detailed family histories were available.
- Nonhematologic phenotypes of GATA2 deficiency were present in 51% of GATA2-mutated patients with MDS and included deafness (9%), lymphedema/hydrocele (23%), and immunodeficiency (39%).
SAMD9 and SAMD9L germline mutations are both associated with pediatric MDS cases in which there is an additional loss of all or part of chromosome 7.[12,13]
In 2016, SAMD9 was identified as the cause of the MIRAGE syndrome (myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy), which is associated with early-onset MDS with monosomy 7.[14] Subsequently, mutations in SAMD9L were identified in patients with ataxia pancytopenia syndrome (ATXPC; OMIM 159550). SAMD9 and SAMD9L mutations were also identified as the cause of myelodysplasia and leukemia syndrome with monosomy 7 (MLSM7; OMIM 252270),[15] a syndrome first identified in phenotypically normal siblings who developed MDS or AML associated with monosomy 7 during childhood.[16]
- Causative mutations in both SAMD9 and SAMD9L are gain-of-function mutations and enhance the growth-suppressing activity of SAMD9 and SAMD9L.[14,16]
- Both SAMD9 and SAMD9L are located at chromosome 7q21.2. Cases of MDS in patients with SAMD9 or SAMD9L mutations often show monosomy 7, with the remaining chromosome 7 having wild-type SAMD9 and SAMD9L. This results in the loss of the enhanced growth-suppressing activity of the mutated gene.
- Phenotypically normal patients with SAMD9 or SAMD9L mutations and monosomy 7 may progress to MDS or AML or, alternatively, may show loss of their monosomy 7 with a return of normal hematopoiesis.[16] The former outcome is associated with the acquisition of mutations in genes associated with MDS/AML (e.g., ETV6 or SETBP1), while the latter is associated with genetic alterations (e.g., revertant mutations or copy-neutral loss of heterozygosity with retention of the wild-type allele) that result in normalization of SAMD9 or SAMD9L activity. These observations suggest that monitoring of patients with SAMD9- or SAMD9L-related monosomy 7 using clinical sequencing for acquired mutations in genes associated with progression to AML may identify patients at high risk of leukemic transformation who may benefit most from hematopoietic stem cell transplantation.[16]
The presence of an isolated monosomy 7 is the most common cytogenetic abnormality, although it does not appear to portend a poor prognosis compared with its presence in overt AML. However, the presence of monosomy 7 in combination with other cytogenetic abnormalities is associated with a poor prognosis.[24,25] The relatively common abnormalities of -Y, 20q-, and 5q- in adults with MDS are rare in childhood MDS. The presence of cytogenetic abnormalities that are found in AML (t(8;21)(q22;q22.1), inv(16)(p13.1;q22) or t(16;16)(p13.1;q22), and APL with PML::RARA gene fusions) defines disease that should be treated as AML and not MDS, regardless of blast percentage. The WHO notes that whether this should also apply to other recurring genetic abnormalities remains controversial.[26]
For more information about the World Health Organization (WHO) classification system of MDS, see the WHO Classification of Bone Marrow and Peripheral Blood Findings for Myelodysplastic Syndromes section.
Classification of MDS
The French-American-British (FAB) and WHO classification systems of MDS and MPS have been difficult to apply to pediatric patients. Alternative classification systems for children have been proposed, but none have been uniformly adopted, with the exception of the modified 2008 WHO classification system.[27,28,29,30,31] The WHO system [32] has been modified for pediatrics.[30] For information about the WHO classification schema and diagnostic criteria, see Table 3 and Table 4. The 2016 revisions to the WHO MDS classification system did not affect classification in children.[33]
The refractory cytopenia subtype represents approximately 50% of all childhood cases of MDS. The presence of an isolated monosomy 7 is the most common cytogenetic abnormality, although it does not appear to portend a poor prognosis compared with its presence in overt AML. However, the presence of monosomy 7 in combination with other cytogenetic abnormalities is associated with a poor prognosis.[24,25] In one retrospective analysis, only the revised International Prognostic Scoring System (R-IPSS) very poor–risk subgroup, defined as having complex cytogenetics (i.e., >3 abnormalities), was found to have a significant adverse prognostic impact on OS and relapse risk after transplant.[34] The relatively common abnormalities of -Y, 20q-, and 5q- in adults with MDS are rare in childhood MDS. The presence of cytogenetic abnormalities that are found in AML (t(8;21)(q22;q22.1), inv(16)(p13.1;q22) or t(16;16)(p13.1;q22), and APL with PML::RARA gene fusions) defines disease that should be treated as AML and not MDS, regardless of blast percentage. The WHO notes that whether this should also apply to other recurring genetic abnormalities remains controversial.[26]
The R-IPSS prognostic groups and associated cytogenetic abnormalities include the following:[34]
- Very good prognostic group: -Y; del(11q).
- Good prognostic group: Normal; del(5q); del(20q); del(12p); double including del(5q).
- Intermediate prognostic group: del(7q); +8; i(17q); +19; any other single or double independent clones.
- Poor prognostic group: -7; inv(3)/t(3q)/del(3q); double including -7/del(7q); complex: 3 abnormalities.
- Very poor prognostic group: Complex: >3 abnormalities.
The IPSS can help to distinguish low-risk from high-risk MDS, although its utility in children with MDS is more limited than in adults because many characteristics differ between children and adults.[35,36] The median survival for children with high-risk MDS remains substantially better than adults, and the presence of monosomy 7 in children has not had the same adverse prognostic impact as does the presence in adults with MDS.[37]
Treatment of Childhood MDS
Treatment options for children with MDS include the following:
- HSCT.
- Other therapies.
HSCT
MDS and associated disorders usually involve a primitive hematopoietic stem cell. Thus, allogeneic HSCT is considered to be the optimal approach to treatment for pediatric patients with MDS. Although matched sibling transplantation is preferred, similar survival has been noted with well-matched, unrelated cord blood and haploidentical approaches.[38,39,40,41,42]
When making treatment decisions, some data should be considered. For example, survival as high as 80% has been reported for patients with early-stage MDS who proceeded to transplant within a few months of diagnosis. Additionally, early transplant and not receiving pretransplant chemotherapy have been associated with improved survival in children with MDS.[43][Level of evidence C1] Disease-free survival (DFS) rates have been estimated to be between 50% to 70% for pediatric patients with advanced MDS using myeloablative transplant preparative regimens.[41,44,45,46,47] While nonmyeloablative preparative transplant regimens are being tested in patients with MDS and AML, such regimens are still investigational for children with these disorders, but may be reasonable in the setting of a clinical trial or when a patient's organ function is compromised in such a way that they would not tolerate a myeloablative regimen.[48,49,50,51]; [52][Level of evidence C1]
The question of whether chemotherapy should be used in high-risk MDS has been examined.
Evidence (HSCT):
- An analysis of 37 children with MDS treated on Berlin-Frankfurt-Münster AML protocols 83, 87, and 93 confirmed the induction response of 74% for patients with refractory anemia with excess blasts in transformation and suggested that transplantation was beneficial.[53]
- Another study by the same group showed that with current approaches to HSCT, survival occurred in more than 60% of children with advanced MDS, and outcomes for patients receiving unrelated donor cells were similar to those for patients who received matched-family donor (MFD) cells.[54]
- The Children's Cancer Group 2891 trial accrued patients between 1989 and 1995, including children with MDS.[44] There were 77 patients enrolled, including patients with refractory anemia (n = 2), refractory anemia with excess blasts (n = 33), refractory anemia with excess blasts in transformation (n = 26), or AML with antecedent MDS (n = 16). Patients were randomly assigned to receive either standard or intensively timed induction. Subsequently, patients were allocated to allogeneic HSCT if there was a suitable family donor, or randomly assigned to either autologous HSCT or chemotherapy.
- Patients with refractory anemia or refractory anemia with excess blasts had a lower remission rate (45%), and those with refractory anemia with excess blasts in transformation (69%) or AML with history of MDS (81%) had similar remission rates compared with de novo AML (77%).
- The 6-year survival rates were lower for those with refractory anemia or refractory anemia with excess blasts (28%) and refractory anemia with excess blasts in transformation (30%).
- Patients with AML and antecedent MDS had a similar outcome to those with de novo AML (50% survival compared with 45%).
- Allogeneic HSCT appeared to improve survival (P = .08).
When analyzing these results, it is important to consider that the subtype refractory anemia with excess blasts in transformation is likely to represent patients with overt AML, while refractory anemia and refractory anemia with excess blasts represents MDS. The WHO classification has now omitted the category of refractory anemia with excess blasts in transformation, concluding that this entity was essentially AML.
Because survival after HSCT is improved in children with early forms of MDS (refractory anemia), transplantation before progression to late MDS or AML should be considered. HSCT should especially be considered when transfusions or other treatment are required, as is usually the case in patients with severe symptomatic cytopenias.[41,47] The 8-year disease-free survival (DFS) rates for children with various stages of MDS has been reported to be 65% for those treated with HLA matched donor transplants and 40% for those treated with mismatched unrelated donor transplants.[47][Level of evidence C2] A 3-year DFS rate of 50% was reported with the use of unrelated cord blood donor transplants for children with MDS, when the transplants were done after the year 2001.[55][Level of evidence C2]
Because MDS in children is often associated with inherited predisposition syndromes, reports of transplantation in small numbers of patients with these disorders have been documented. For example, in patients with Fanconi anemia and AML or advanced MDS, the 5-year overall survival (OS) rate has been reported to be 33% to 55%.[56,57][Level of evidence C1] Second transplants have also been used in pediatric patients with MDS/MPD who relapse or suffer graft failure. The 3-year OS rates were 33% for those retransplanted after relapse and 57% for those transplanted after initial graft failure.[58][Level of evidence C1]
While some patients with inherited predisposition syndromes require significant modification of their transplant approaches because of excess toxicity (e.g., Fanconi anemia), other syndromes have no detectable excessive toxicity associated with the transplant process. Inherited GATA2 is a good example of the latter. One study compared HSCT outcomes of 65 children with GATA2 germline mutations and MDS with the outcomes of 404 children with MDS and wild-type germline GATA2. Disease-free survival, relapse, and nonrelapse mortality were similar in the two populations.[59]
For patients with clinically significant cytopenias, supportive care that includes transfusions and prophylactic antibiotics are considered standard of care. The use of hematopoietic growth factors can improve the hematopoietic status, but concerns remain that such treatment could accelerate conversion to AML.[60]
Other therapies
Other supportive therapies that have been studied include the following:
- Steroid therapies, including glucocorticoids and androgens, have been tested with mixed results.[61]
- Treatments directed toward scavenging free oxygen radicals with amifostine [62,63] or the use of differentiation-promoting retinoids,[64] DNA methylation inhibitors (e.g., azacitidine and decitabine), and histone deacetylase inhibitors have all shown some response, but no definitive trials in children with MDS have been reported. Azacitidine has been approved by the U.S. Food and Drug Administration (FDA) for the treatment of MDS in adults on the basis of randomized studies.[65] For more information, see the Disease-Modifying Agents section in Myelodysplastic Syndromes Treatment.
- Agents such as lenalidomide, an analog of thalidomide, have been tested based on findings that demonstrated increased activity in the bone marrow of patients with MDS. Lenalidomide has shown the most efficacy in patients with 5q- syndrome, especially those with thrombocytosis, and is now FDA approved for use in adults with this finding.[66]
- Immunosuppression with antithymocyte globulin and/or cyclosporine has also been reported in adults.[66,67]
Treatment Options Under Clinical Evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Alter BP, Giri N, Savage SA, et al.: Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol 150 (2): 179-88, 2010.
- Rosenberg PS, Huang Y, Alter BP: Individualized risks of first adverse events in patients with Fanconi anemia. Blood 104 (2): 350-5, 2004.
- Ludwig LS, Gazda HT, Eng JC, et al.: Altered translation of GATA1 in Diamond-Blackfan anemia. Nat Med 20 (7): 748-53, 2014.
- Rosenberg PS, Zeidler C, Bolyard AA, et al.: Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br J Haematol 150 (2): 196-9, 2010.
- Wechsler J, Greene M, McDevitt MA, et al.: Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 32 (1): 148-52, 2002.
- Liew E, Owen C: Familial myelodysplastic syndromes: a review of the literature. Haematologica 96 (10): 1536-42, 2011.
- Owen C, Barnett M, Fitzgibbon J: Familial myelodysplasia and acute myeloid leukaemia--a review. Br J Haematol 140 (2): 123-32, 2008.
- Ghauri RI, Naveed M, Mannan J: Congenital amegakaryocytic thrombocytopenic purpura (CAMT). J Coll Physicians Surg Pak 24 (4): 285-7, 2014.
- Auer PL, Teumer A, Schick U, et al.: Rare and low-frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat Genet 46 (6): 629-34, 2014.
- Vinh DC, Patel SY, Uzel G, et al.: Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood 115 (8): 1519-29, 2010.
- Schwartz JR, Ma J, Lamprecht T, et al.: The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun 8 (1): 1557, 2017.
- Schwartz JR, Wang S, Ma J, et al.: Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia 31 (8): 1827-1830, 2017.
- Davidsson J, Puschmann A, Tedgård U, et al.: SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 32 (5): 1106-1115, 2018.
- Narumi S, Amano N, Ishii T, et al.: SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet 48 (7): 792-7, 2016.
- Chen DH, Below JE, Shimamura A, et al.: Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am J Hum Genet 98 (6): 1146-1158, 2016.
- Wong JC, Bryant V, Lamprecht T, et al.: Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight 3 (14): , 2018.
- Keel SB, Scott A, Sanchez-Bonilla M, et al.: Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients. Haematologica 101 (11): 1343-1350, 2016.
- Kasahara S, Hara T, Itoh H, et al.: Hypoplastic myelodysplastic syndromes can be distinguished from acquired aplastic anaemia by bone marrow stem cell expression of the tumour necrosis factor receptor. Br J Haematol 118 (1): 181-8, 2002.
- Orazi A: Histopathology in the diagnosis and classification of acute myeloid leukemia, myelodysplastic syndromes, and myelodysplastic/myeloproliferative diseases. Pathobiology 74 (2): 97-114, 2007.
- Pastor V, Hirabayashi S, Karow A, et al.: Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia 31 (3): 759-762, 2017.
- Collin M, Dickinson R, Bigley V: Haematopoietic and immune defects associated with GATA2 mutation. Br J Haematol 169 (2): 173-87, 2015.
- Wlodarski MW, Hirabayashi S, Pastor V, et al.: Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 127 (11): 1387-97; quiz 1518, 2016.
- Wlodarski MW, Collin M, Horwitz MS: GATA2 deficiency and related myeloid neoplasms. Semin Hematol 54 (2): 81-86, 2017.
- Göhring G, Michalova K, Beverloo HB, et al.: Complex karyotype newly defined: the strongest prognostic factor in advanced childhood myelodysplastic syndrome. Blood 116 (19): 3766-9, 2010.
- Haase D, Germing U, Schanz J, et al.: New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood 110 (13): 4385-95, 2007.
- Arber DA, Vardiman JW, Brunning RD: Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 110-23.
- Occhipinti E, Correa H, Yu L, et al.: Comparison of two new classifications for pediatric myelodysplastic and myeloproliferative disorders. Pediatr Blood Cancer 44 (3): 240-4, 2005.
- Niemeyer CM, Baumann I: Myelodysplastic syndrome in children and adolescents. Semin Hematol 45 (1): 60-70, 2008.
- Niemeyer CM, Kratz CP: Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classification and treatment options. Br J Haematol 140 (6): 610-24, 2008.
- Hasle H: Myelodysplastic and myeloproliferative disorders in children. Curr Opin Pediatr 19 (1): 1-8, 2007.
- Mandel K, Dror Y, Poon A, et al.: A practical, comprehensive classification for pediatric myelodysplastic syndromes: the CCC system. J Pediatr Hematol Oncol 24 (7): 596-605, 2002.
- Nösslinger T, Reisner R, Koller E, et al.: Myelodysplastic syndromes, from French-American-British to World Health Organization: comparison of classifications on 431 unselected patients from a single institution. Blood 98 (10): 2935-41, 2001.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Yamamoto S, Kato M, Watanabe K, et al.: Prognostic value of the revised International Prognostic Scoring System five-group cytogenetic abnormality classification for the outcome prediction of hematopoietic stem cell transplantation in pediatric myelodysplastic syndrome. Bone Marrow Transplant 56 (12): 3016-3023, 2021.
- Cutler CS, Lee SJ, Greenberg P, et al.: A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood 104 (2): 579-85, 2004.
- Hasle H, Baumann I, Bergsträsser E, et al.: The International Prognostic Scoring System (IPSS) for childhood myelodysplastic syndrome (MDS) and juvenile myelomonocytic leukemia (JMML). Leukemia 18 (12): 2008-14, 2004.
- Hasle H, Niemeyer CM: Advances in the prognostication and management of advanced MDS in children. Br J Haematol 154 (2): 185-95, 2011.
- Uberti JP, Agovi MA, Tarima S, et al.: Comparative analysis of BU and CY versus CY and TBI in full intensity unrelated marrow donor transplantation for AML, CML and myelodysplasia. Bone Marrow Transplant 46 (1): 34-43, 2011.
- Nemecek ER, Guthrie KA, Sorror ML, et al.: Conditioning with treosulfan and fludarabine followed by allogeneic hematopoietic cell transplantation for high-risk hematologic malignancies. Biol Blood Marrow Transplant 17 (3): 341-50, 2011.
- Shaw PJ, Kan F, Woo Ahn K, et al.: Outcomes of pediatric bone marrow transplantation for leukemia and myelodysplasia using matched sibling, mismatched related, or matched unrelated donors. Blood 116 (19): 4007-15, 2010.
- Parikh SH, Mendizabal A, Martin PL, et al.: Unrelated donor umbilical cord blood transplantation in pediatric myelodysplastic syndrome: a single-center experience. Biol Blood Marrow Transplant 15 (8): 948-55, 2009.
- Locatelli F, Merli P, Pagliara D, et al.: Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood 130 (5): 677-685, 2017.
- Smith AR, Christiansen EC, Wagner JE, et al.: Early hematopoietic stem cell transplant is associated with favorable outcomes in children with MDS. Pediatr Blood Cancer 60 (4): 705-10, 2013.
- Woods WG, Barnard DR, Alonzo TA, et al.: Prospective study of 90 children requiring treatment for juvenile myelomonocytic leukemia or myelodysplastic syndrome: a report from the Children's Cancer Group. J Clin Oncol 20 (2): 434-40, 2002.
- Andolina JR, Kletzel M, Tse WT, et al.: Allogeneic hematopoetic stem cell transplantation in pediatric myelodysplastic syndromes: improved outcomes for de novo disease. Pediatr Transplant 15 (3): 334-43, 2011.
- Al-Seraihy A, Ayas M, Al-Nounou R, et al.: Outcome of allogeneic stem cell transplantation with a conditioning regimen of busulfan, cyclophosphamide and low-dose etoposide for children with myelodysplastic syndrome. Hematol Oncol Stem Cell Ther 4 (3): 121-5, 2011.
- Woodard P, Carpenter PA, Davies SM, et al.: Unrelated donor bone marrow transplantation for myelodysplastic syndrome in children. Biol Blood Marrow Transplant 17 (5): 723-8, 2011.
- Champlin R: Hematopoietic stem cell transplantation for treatment of myleodysplastic syndromes. Biol Blood Marrow Transplant 17 (1 Suppl): S6-8, 2011.
- Nelson RP, Yu M, Schwartz JE, et al.: Long-term disease-free survival after nonmyeloablative cyclophosphamide/fludarabine conditioning and related/unrelated allotransplantation for acute myeloid leukemia/myelodysplasia. Bone Marrow Transplant 45 (8): 1300-8, 2010.
- Warlick ED: Optimizing stem cell transplantation in myelodysplastic syndromes: unresolved questions. Curr Opin Oncol 22 (2): 150-4, 2010.
- Pulsipher MA, Boucher KM, Wall D, et al.: Reduced-intensity allogeneic transplantation in pediatric patients ineligible for myeloablative therapy: results of the Pediatric Blood and Marrow Transplant Consortium Study ONC0313. Blood 114 (7): 1429-36, 2009.
- Gao L, Gao L, Gong Y, et al.: Reduced-intensity conditioning therapy with fludarabine, idarubicin, busulfan and cytarabine for allogeneic hematopoietic stem cell transplantation in acute myeloid leukemia and myelodysplastic syndrome. Leuk Res 37 (11): 1482-7, 2013.
- Creutzig U, Bender-Götze C, Ritter J, et al.: The role of intensive AML-specific therapy in treatment of children with RAEB and RAEB-t. Leukemia 12 (5): 652-9, 1998.
- Strahm B, Nöllke P, Zecca M, et al.: Hematopoietic stem cell transplantation for advanced myelodysplastic syndrome in children: results of the EWOG-MDS 98 study. Leukemia 25 (3): 455-62, 2011.
- Madureira AB, Eapen M, Locatelli F, et al.: Analysis of risk factors influencing outcome in children with myelodysplastic syndrome after unrelated cord blood transplantation. Leukemia 25 (3): 449-54, 2011.
- Mitchell R, Wagner JE, Hirsch B, et al.: Haematopoietic cell transplantation for acute leukaemia and advanced myelodysplastic syndrome in Fanconi anaemia. Br J Haematol 164 (3): 384-95, 2014.
- Ayas M, Saber W, Davies SM, et al.: Allogeneic hematopoietic cell transplantation for fanconi anemia in patients with pretransplantation cytogenetic abnormalities, myelodysplastic syndrome, or acute leukemia. J Clin Oncol 31 (13): 1669-76, 2013.
- Kato M, Yoshida N, Inagaki J, et al.: Salvage allogeneic stem cell transplantation in patients with pediatric myelodysplastic syndrome and myeloproliferative neoplasms. Pediatr Blood Cancer 61 (10): 1860-6, 2014.
- Bortnick R, Wlodarski M, de Haas V, et al.: Hematopoietic stem cell transplantation in children and adolescents with GATA2-related myelodysplastic syndrome. Bone Marrow Transplant 56 (11): 2732-2741, 2021.
- Zwierzina H, Suciu S, Loeffler-Ragg J, et al.: Low-dose cytosine arabinoside (LD-AraC) vs LD-AraC plus granulocyte/macrophage colony stimulating factor vs LD-AraC plus Interleukin-3 for myelodysplastic syndrome patients with a high risk of developing acute leukemia: final results of a randomized phase III study (06903) of the EORTC Leukemia Cooperative Group. Leukemia 19 (11): 1929-33, 2005.
- Chan G, DiVenuti G, Miller K: Danazol for the treatment of thrombocytopenia in patients with myelodysplastic syndrome. Am J Hematol 71 (3): 166-71, 2002.
- Mathew P, Gerbing R, Alonzo TA, et al.: A phase II study of amifostine in children with myelodysplastic syndrome: a report from the Children's Oncology Group study (AAML0121). Pediatr Blood Cancer 57 (7): 1230-2, 2011.
- Schanz J, Jung H, Wörmann B, et al.: Amifostine has the potential to induce haematologic responses and decelerate disease progression in individual patients with low- and intermediate-1-risk myelodysplastic syndromes. Leuk Res 33 (9): 1183-8, 2009.
- Sadek I, Zayed E, Hayne O, et al.: Prolonged complete remission of myelodysplastic syndrome treated with danazol, retinoic acid and low-dose prednisone. Am J Hematol 64 (4): 306-10, 2000.
- Silverman LR, Demakos EP, Peterson BL, et al.: Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 20 (10): 2429-40, 2002.
- Yazji S, Giles FJ, Tsimberidou AM, et al.: Antithymocyte globulin (ATG)-based therapy in patients with myelodysplastic syndromes. Leukemia 17 (11): 2101-6, 2003.
- Yoshimi A, Baumann I, Führer M, et al.: Immunosuppressive therapy with anti-thymocyte globulin and cyclosporine A in selected children with hypoplastic refractory cytopenia. Haematologica 92 (3): 397-400, 2007.
Therapy-Related AML and Therapy-Related Myelodysplastic Syndromes
Pathogenesis
The development of acute myeloid leukemia (AML) or myelodysplastic syndromes (MDS) after treatment with ionizing radiation or chemotherapy, particularly alkylating agents and topoisomerase inhibitors, is termed therapy-related AML (t-AML) or therapy-related MDS (t-MDS). In addition to genotoxic exposures, genetic predisposition susceptibilities (such as polymorphisms in drug detoxification and DNA repair pathway components) may contribute to the occurrence of secondary AML/MDS.[1,2,3,4]
The risk of t-AML or t-MDS is regimen-dependent and often related to the cumulative doses of chemotherapy agents received and the dose and field of radiation administered.[5] Regimens previously used that employed high cumulative doses of either epipodophyllotoxins (e.g., etoposide or teniposide) or alkylating agents (e.g., mechlorethamine, melphalan, busulfan, and cyclophosphamide) induced excessively high rates of t-AML or t-MDS that exceeded 10% in some cases.[5,6] However, most current chemotherapy regimens that are used to treat childhood cancers have a cumulative incidence of t-AML or t-MDS no greater than 1% to 2%.
t-AML or t-MDS resulting from epipodophyllotoxins and other topoisomerase II inhibitors (e.g., anthracyclines) usually occur within 2 years of exposure and are commonly associated with chromosome 11q23 abnormalities,[7] although other subtypes of AML (e.g., acute promyelocytic leukemia) have been reported.[8,9] t-AML that occurs after exposure to alkylating agents or ionizing radiation often presents 5 to 7 years later and is commonly associated with monosomies or deletions of chromosomes 5 and 7.[1,7]
Treatment of t-AML or t-MDS
Treatment options for t-AML or t-MDS include the following:
- Hematopoietic stem cell transplantation (HSCT).
The goal of treatment is to achieve an initial complete remission (CR) using AML-directed regimens and then, usually, to proceed directly to HSCT with the best available donor. However, treatment is challenging because of the following:[10]
- Increased rates of adverse cytogenetics and subsequent failure to obtain remission with chemotherapy.
- Comorbidities or limitations related to chemotherapy for the previous malignancy.
Accordingly, CR rates and overall survival (OS) rates are usually lower for patients with t-AML compared with patients with de novo AML.[10,11,12] Also, survival for pediatric patients with t-MDS is worse than survival for pediatric patients with MDS not related to previous therapy.[13]
Patients with t-MDS-refractory anemia usually have not needed induction chemotherapy before transplant; the role of induction therapy before transplant is controversial in patients with refractory anemia with excess blasts-1.
Only a few reports describe the outcome of children undergoing HSCT for t-AML.
Evidence (HSCT for t-AML or t-MDS):
- One study described the outcomes of 27 children with t-AML who received related and unrelated donor HSCT.[14]
- Three-year OS rates were 18.5% (± 7.5%) and event-free survival (EFS) rates were 18.7% (± 7.5%).
- Poor survival was mainly the result of very high transplant-related mortality (59.6% ± 8.4%).
- Another study reported a second retrospective single-center experience of 14 patients with t-AML or t-MDS who underwent transplantation between 1975 and 2007.[11]
- Survival was 29%, but in this review, only 63% of patients diagnosed with t-AML or t-MDS underwent HSCT.
- A multicenter study (CCG-2891) examined outcomes of 24 children with t-AML or t-MDS compared with other children enrolled on the study with de novo AML (n = 898) or MDS (n = 62). Children with t-AML or t-MDS were older and low-risk cytogenetics rarely occurred.[15]
- Although rates of achieving CR and OS at 3 years were worse in the t-AML/t-MDS group (CR, 50% vs. 72%; P = .016; OS rate, 26% vs. 47%; P = .007), survival was similar (OS rate, 45% vs. 53%; P = .87) if patients achieved a CR.
- The importance of remission to survival in these patients is further illustrated by another single-center report of 21 children who underwent HSCT for t-AML or t-MDS between 1994 and 2009. Of the 21 children, 12 had t-AML (11 in CR at the time of transplant), seven had refractory anemia (for whom induction was not done), and two had refractory anemia with excess blasts.[16]
- The survival rate of the entire cohort was 61%. Patients in remission or with refractory anemia had a disease-free survival rate of 66%; for the three patients with more than 5% blasts at the time of HSCT, the survival rate was 0% (P = .015).
Because t-AML is rare in children, it is not known whether the significant decrease in transplant-related mortality after unrelated donor HSCT noted over the past several years will translate to improved survival in this population. Patients should be carefully assessed for pre-HSCT morbidities caused by earlier therapies, and treatment approaches should be adapted to give adequate intensity while minimizing transplant-related mortality.
References:
- Leone G, Fianchi L, Voso MT: Therapy-related myeloid neoplasms. Curr Opin Oncol 23 (6): 672-80, 2011.
- Bolufer P, Collado M, Barragan E, et al.: Profile of polymorphisms of drug-metabolising enzymes and the risk of therapy-related leukaemia. Br J Haematol 136 (4): 590-6, 2007.
- Ezoe S: Secondary leukemia associated with the anti-cancer agent, etoposide, a topoisomerase II inhibitor. Int J Environ Res Public Health 9 (7): 2444-53, 2012.
- Ding Y, Sun CL, Li L, et al.: Genetic susceptibility to therapy-related leukemia after Hodgkin lymphoma or non-Hodgkin lymphoma: role of drug metabolism, apoptosis and DNA repair. Blood Cancer J 2 (3): e58, 2012.
- Leone G, Mele L, Pulsoni A, et al.: The incidence of secondary leukemias. Haematologica 84 (10): 937-45, 1999.
- Pui CH, Ribeiro RC, Hancock ML, et al.: Acute myeloid leukemia in children treated with epipodophyllotoxins for acute lymphoblastic leukemia. N Engl J Med 325 (24): 1682-7, 1991.
- Andersen MK, Johansson B, Larsen SO, et al.: Chromosomal abnormalities in secondary MDS and AML. Relationship to drugs and radiation with specific emphasis on the balanced rearrangements. Haematologica 83 (6): 483-8, 1998.
- Ogami A, Morimoto A, Hibi S, et al.: Secondary acute promyelocytic leukemia following chemotherapy for non-Hodgkin's lymphoma in a child. J Pediatr Hematol Oncol 26 (7): 427-30, 2004.
- Okamoto T, Okada M, Wakae T, et al.: Secondary acute promyelocytic leukemia in a patient with non-Hodgkin's lymphoma treated with VP-16 and MST-16. Int J Hematol 75 (1): 107-8, 2002.
- Larson RA: Etiology and management of therapy-related myeloid leukemia. Hematology Am Soc Hematol Educ Program : 453-9, 2007.
- Aguilera DG, Vaklavas C, Tsimberidou AM, et al.: Pediatric therapy-related myelodysplastic syndrome/acute myeloid leukemia: the MD Anderson Cancer Center experience. J Pediatr Hematol Oncol 31 (11): 803-11, 2009.
- Yokoyama H, Mori S, Kobayashi Y, et al.: Hematopoietic stem cell transplantation for therapy-related myelodysplastic syndrome and acute leukemia: a single-center analysis of 47 patients. Int J Hematol 92 (2): 334-41, 2010.
- Xavier AC, Kutny M, Costa LJ: Incidence and outcomes of paediatric myelodysplastic syndrome in the United States. Br J Haematol 180 (6): 898-901, 2018.
- Woodard P, Barfield R, Hale G, et al.: Outcome of hematopoietic stem cell transplantation for pediatric patients with therapy-related acute myeloid leukemia or myelodysplastic syndrome. Pediatr Blood Cancer 47 (7): 931-5, 2006.
- Barnard DR, Lange B, Alonzo TA, et al.: Acute myeloid leukemia and myelodysplastic syndrome in children treated for cancer: comparison with primary presentation. Blood 100 (2): 427-34, 2002.
- Kobos R, Steinherz PG, Kernan NA, et al.: Allogeneic hematopoietic stem cell transplantation for pediatric patients with treatment-related myelodysplastic syndrome or acute myelogenous leukemia. Biol Blood Marrow Transplant 18 (3): 473-80, 2012.
Juvenile Myelomonocytic Leukemia (JMML)
Incidence
Juvenile myelomonocytic leukemia (JMML) is a rare leukemia that occurs approximately ten times less frequently than acute myeloid leukemia (AML) in children, with an annual incidence of about 1 to 2 cases per 1 million people.[1] JMML typically presents in young children (median age, approximately 1.8 years) and occurs more commonly in boys (male to female ratio, approximately 2.5:1).
Clinical Presentation and Diagnostic Criteria
Common clinical features at diagnosis include the following:[2]
- Hepatosplenomegaly (97%).
- Lymphadenopathy (76%).
- Pallor (64%).
- Fever (54%).
- Skin rash (36%).
In children presenting with clinical features suggestive of JMML, current criteria used for a definitive diagnosis are described in Table 8.[3]
| Category 1 (All are Required) | Category 2 (One is Sufficient)a | Category 3 (Patients Without Genetic Features Must Have the Following in Addition to Category 1b) |
|---|---|---|
| Clinical and Hematologic Features | Genetic Studies | Other Features |
| GM-CSF = granulocyte-macrophage colony-stimulating factor; NF1 = neurofibromatosis type 1. | ||
| a Patients who are found to have a category 2 lesion need to meet the criteria in category 1 but do not need to meet the category 3 criteria. Patients who are not found to have a category 2 lesion must meet the category 1 and 3 criteria. | ||
| b Note that only 7% of patients with JMML will NOT present with splenomegaly, but virtually all patients develop splenomegaly within several weeks to months of initial presentation. | ||
| Absence of theBCR::ABL1gene fusion | Somatic mutation inKRAS,NRAS, orPTPN11(germline mutations need to be excluded) | Monosomy 7 or other chromosomal abnormality, or at least 2 of the criteria listed below: |
| >1 × 109 /L circulating monocytes | Clinical diagnosis of NF1 orNF1gene mutation | — Circulating myeloid or erythroid precursors |
| <20% blasts in the peripheral blood and bone marrow | GermlineCBLmutation and loss of heterozygosity ofCBL | — Increased hemoglobin F for age |
| Splenomegaly | — Hyperphosphorylation of STAT5 | |
| — GM-CSF hypersensitivity | ||
Pathogenesis and Related Syndromes
The pathogenesis of JMML has been closely linked to activation of the RAS oncogene pathway, along with related syndromes (see Figure 1).[4,5] In addition, distinctive RNA expression and DNA methylation patterns have been reported; they are correlated with clinical factors such as age and appear to be associated with prognosis.[6,7]
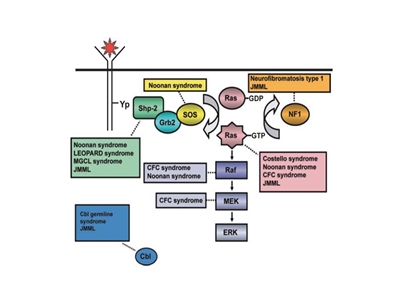
Figure 1. Schematic diagram showing ligand-stimulated Ras activation, the Ras-Erk pathway, and the gene mutations found to date contributing to the neuro-cardio-facio-cutaneous congenital disorders and JMML. NL/MGCL: Noonan-like/multiple giant cell lesion; CFC: cardia-facio-cutaneous; JMML: juvenile myelomonocytic leukemia. Reprinted from Leukemia Research, 33 (3), Rebecca J. Chan, Todd Cooper, Christian P. Kratz, Brian Weiss, Mignon L. Loh, Juvenile myelomonocytic leukemia: A report from the 2nd International JMML Symposium, Pages 355-62, Copyright 2009, with permission from Elsevier.
Children with neurofibromatosis type 1 (NF1) and Noonan syndrome are at increased risk of developing JMML:[8,9]
- NF1. Up to 14% of cases of JMML occur in children with NF1.[2]
- Noonan syndrome. Noonan syndrome is usually inherited as an autosomal dominant condition but can also arise spontaneously. It is characterized by facial dysmorphism, short stature, webbed neck, neurocognitive abnormalities, and cardiac abnormalities. Germline mutations in PTPN11 are observed in children with Noonan syndrome and in children with JMML.[10,11,12]
Importantly, some children with Noonan syndrome have a hematologic picture indistinguishable from JMML that self-resolves during infancy, similar to what happens in children with Down syndrome and transient myeloproliferative disorder.[5,12]
Within a large prospective cohort of 641 patients with Noonan syndrome and a germline PTPN11 mutation, 36 patients (approximately 6%) showed myeloproliferative features, with 20 patients (approximately 3%) meeting the consensus diagnostic criteria for JMML.[12] Of the 20 patients meeting the criteria for JMML, 12 patients had severe neonatal manifestations (e.g., life-threatening complications related to congenital heart defects, pleural effusion, leukemia infiltrates, and/or thrombocytopenia), and 10 of 20 patients died during the first month of life. Among the remaining eight patients, none required intensive therapy at diagnosis or during follow-up. All 16 patients with myeloproliferative features that did not meet JMML criteria were alive, with a median follow-up of 3 years, and none of the patients received chemotherapy.
Mutations in the CBL gene, an E3 ubiquitin-protein ligase that is involved in targeting proteins, particularly tyrosine kinases, for proteasomal degradation occur in 10% to 15% of JMML cases,[13,14] with many of these cases occurring in children with germline CBL mutations.[15,16,17]CBL germline mutations result in an autosomal dominant developmental disorder that is often characterized by impaired growth, developmental delay, cryptorchidism, and a predisposition to JMML.[15,17] Some individuals with CBL germline mutations experience spontaneous regression of their JMML but develop vasculitis later in life,[15] whereas patients with only somatic CBL mutations all require therapy.[17] JMML arising from germline mutations are clinically indistinguishable from JMML arising from somatic mutations, which necessitates studies on both normal and leukemic tissue.[17]CBL mutations are nearly always mutually exclusive of RAS and PTPN11 mutations.[13]
Genomics of JMML
Molecular features of JMML
The genomic landscape of JMML is characterized by mutations in one of five genes of the RAS pathway: NF1, NRAS, KRAS, PTPN11, and CBL.[18,19,20] In a series of 118 consecutively diagnosed JMML cases with RAS pathway–activating mutations, PTPN11 was the most commonly mutated gene, accounting for 51% of cases (19% germline and 32% somatic) (see Figure 2).[18] Patients with mutated NRAS accounted for 19% of cases, and patients with mutated KRAS accounted for 15% of cases. NF1 mutations accounted for 8% of cases, and CBL mutations accounted for 11% of cases. Although mutations among these five genes are generally mutually exclusive, 4% to 17% of cases have mutations in two of these RAS pathway genes,[18,19,20] a finding that is associated with poorer prognosis.[18,20]
The mutation rate in JMML leukemia cells is very low, but additional mutations beyond those of the five RAS pathway genes described above are observed.[18,19,20] Secondary genomic alterations are observed for genes of the transcriptional repressor complex PRC2 (e.g., ASXL1 was mutated in 7%–8% of cases). Some genes associated with myeloproliferative neoplasms in adults are also mutated at low rates in JMML (e.g., SETBP1 was mutated in 6%–9% of cases).[18,19,20,21]JAK3 mutations are also observed in a small percentage (4%–12%) of JMML cases.[18,19,20,21] Cases with germline PTPN11 and germline CBL mutations showed low rates of additional mutations (see Figure 2).[18] The presence of mutations beyond disease-defining RAS pathway mutations is associated with an inferior prognosis.[18,19]
A report describing the genomic landscape of JMML found that 16 of 150 patients (11%) lacked canonical RAS pathway mutations. Among these 16 patients, 3 were observed to have in-frame fusions involving receptor tyrosine kinases (DCTN1::ALK, RANBP2::ALK, and TBL1XR1::ROS1 gene fusions). These patients all had monosomy 7 and were aged 56 months or older. One patient with an ALK fusion was treated with crizotinib plus conventional chemotherapy and achieved a complete molecular remission and proceeded to allogeneic bone marrow transplantation.[20]
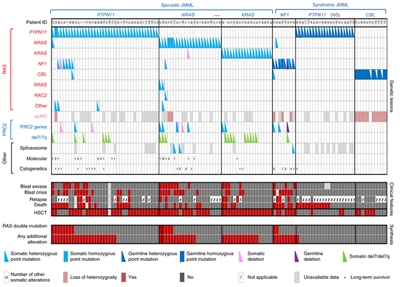
Figure 2. Alteration profiles in individual JMML cases. Germline and somatically acquired alterations with recurring hits in the RAS pathway and PRC2 network are shown for 118 patients with JMML who underwent detailed genetic analysis. Blast excess was defined as a blast count ≥10% but <20% of nucleated cells in the bone marrow at diagnosis. Blast crisis was defined as a blast count ≥20% of nucleated cells in the bone marrow. NS, Noonan syndrome. Reprinted by permission from Macmillan Publishers Ltd: Nature Genetics (Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 [11]: 1334-40, 2015), copyright (2015).
Prognosis (genomic and molecular factors)
Several genomic factors affect the prognosis of patients with JMML, including the following:
- Number of non–RAS pathway mutations. A predictor of prognosis for children with JMML is the number of mutations beyond the disease-defining RAS pathway mutations.[18,19]
- One study observed that zero or one somatic alteration (pathogenic mutation or monosomy 7) was identified in 64 patients (65.3%) at diagnosis, whereas two or more alterations were identified in 34 patients (34.7%).[19] In multivariate analysis, mutation number (2 or more vs. 0 or 1) maintained significance as a predictor of inferior event-free survival (EFS) and overall survival (OS). A higher proportion of patients diagnosed with two or more alterations were older and male, and these patients also demonstrated a higher rate of monosomy 7 or somatic NF1 mutations.[19]
- Another study observed that approximately 60% of patients had one or more additional mutations beyond their disease-defining RAS pathway mutation. These patients had an inferior OS compared with patients who had no additional mutations (3-year OS rate, 61% vs. 85%, respectively).[18]
- A third study observed a trend for an inferior OS for patients with two or more mutations compared with patients with zero or one mutation.[20]
- RAS pathway double mutations. Although mutations in the five canonical RAS pathway genes associated with JMML (NF1, NRAS, KRAS, PTPN11, and CBL) are generally mutually exclusive, 4% to 17% of cases have mutations in two of these RAS pathway genes,[18,19] a finding that has been associated with a poorer prognosis.[18,19]
- Two RAS pathway mutations were identified in 11% of JMML patients in one report, and these patients had a significantly inferior EFS rate (14%) compared with patients who had a single RAS pathway mutation (62%). Patients with Noonan syndrome were excluded from the analyses.[19]
- Similar findings for RAS pathway mutations were reported in a second study that observed that patients with RAS pathway double mutations (15 of 96 patients) had lower survival rates than did patients with either no additional mutations or with additional mutations beyond the RAS pathway mutation.[18]
- DNA methylation profile.
- One study applied DNA methylation profiling to a discovery cohort of 39 patients with JMML and to a validation cohort of 40 patients. Distinctive subsets of JMML with either high, intermediate, or low methylation levels were observed in both cohorts. Patients with the lowest methylation levels had the highest survival rates, and all but 1 of 15 patients experienced spontaneous resolution in the low methylation cohort. High methylation status was associated with lower EFS rates.[22]
- Another study applied DNA methylation profiling to a cohort of 106 patients with JMML and observed one subgroup of patients with a hypermethylation profile and one subgroup of patients with a hypomethylation profile. Patients in the hypermethylation group had a significantly lower OS rate than did patients in the hypomethylation group (5-year OS rate, 46% vs. 73%, respectively). Patients in the hypermethylation group also had a significantly poorer 5-year transplant-free survival rate than did patients in the hypomethylation group (2.2%; 95% CI, 0.2%–10.1% vs. 41.2%; 95% CI, 27.1%–54.8%). Hypermethylation status was associated with two or more mutations, higher fetal hemoglobin levels, older age, and lower platelet count at diagnosis. All patients with Noonan syndrome were in the hypomethylation group.[20]
- A study examined 33 patients with JMML who had CBL mutations and identified 31 patients with low methylation and 2 patients with intermediate methylation. Both of the children with intermediate methylation relapsed after undergoing HSCT. Because treatment, which included observation only, varied among the 31 patients with low methylation, the impact of the methylation profile on therapeutic decisions and outcomes could not be fully assessed. However, the methylation status was not prognostic of spontaneous resolution.[17]
- LIN28B overexpression. LIN28B overexpression is present in approximately one-half of children with JMML and identifies a biologically distinctive subset of JMML. LIN28B is an RNA-binding protein that regulates stem cell renewal.[23]
- LIN28B overexpression was positively correlated with high blood fetal hemoglobin level and age (both of which are associated with poor prognosis), and it was negatively correlated with presence of monosomy 7 (also associated with inferior prognosis). Although LIN28B overexpression identifies a subset of patients with increased risk of treatment failure, it was not found to be an independent prognostic factor when other factors such as age and monosomy 7 status are considered.[23]
- Another study also observed a subset of JMML patients with elevated LIN28B expression and identified LIN28B as the gene for which expression was most strongly associated with hypermethylation status.[20]
Prognosis (Clinical Factors)
Age, platelet count, and fetal hemoglobin level after any treatment. Historically, more than 90% of patients with JMML died despite the use of chemotherapy;[24] however, with the application of hematopoietic stem cell transplantation (HSCT), survival rates of approximately 50% are now observed.[25] Patients appeared to follow three distinct clinical courses:
- Rapidly progressive disease and early demise.
- Transiently stable disease followed by progression and death.
- Clinical improvement that lasted up to 9 years before progression or, rarely, long-term survival.
Favorable prognostic factors for survival after any therapy include age younger than 2 years, platelet count greater than 33 × 109 /L, and low age-adjusted fetal hemoglobin levels.[1,2] In contrast, being older than 2 years and having high blood fetal hemoglobin levels at diagnosis are predictors of poor outcome.[1,2]
Treatment of JMML
Treatment options for JMML include the following:
- Hematopoietic stem cell transplant (HSCT).
The role of conventional antileukemia therapy in the treatment of JMML is not defined. Determining the role of specific agents in the treatment of JMML is complicated because of the absence of consensus response criteria.[26] Some agents that have shown antileukemia activity against JMML include etoposide, cytarabine, thiopurines (thioguanine and mercaptopurine), isotretinoin, and farnesyl inhibitors, but none of these have been shown to improve outcome.[26,27,28,29,30]; [31][Level of evidence B4]
HSCT currently offers the best chance of cure for JMML.[25,32,33,34,35]
Evidence (HSCT):
- A report from the European Working Group on Childhood Myelodysplastic Syndromes included 100 transplant recipients at multiple centers treated with a common preparative regimen of busulfan, cyclophosphamide, and melphalan, with or without antithymocyte globulin. Recipients had been treated with varying degrees of pretransplant chemotherapy or differentiating agents, and some patients had splenectomy performed.[25]
- The 5-year event-free survival rate was 55% for children with JMML who underwent HSCT using HLA-identical matched family donor cells and 49% for children with JMML who underwent HSCT using unrelated donor cells.
- The multivariate analysis showed no effect on survival of previous AML-like chemotherapy versus low-dose chemotherapy or no chemotherapy.
- No effect on survival was observed for splenectomy pretransplant or difference in spleen size.
- Comparison of outcomes on the basis of related versus unrelated donors also found no difference.
- Only age older than 4 years and sex were shown to be poor prognostic factors for outcome and increased risk of relapse (relative risk [RR], 2.24 [1.07–4.69]; P = .032 for older age; RR, 2.22 [1.09–4.50]; P = .028 for females).[25]
- Cord blood transplantation results in a 5-year disease-free survival rate of 44%, with improved outcome in children younger than 1.4 years at diagnosis, those with nonmonosomy 7 karyotype, and those receiving 5/6 to 6/6 HLA-matched cord units.[36][Level of evidence C2] This suggests that cord blood can provide an additional donor pool for this group of children.
- The use of reduced-intensity preparative regimens to decrease the adverse side effects of transplantation have also been reported in small numbers of patients, generally for patients ineligible for myeloablative HSCT.[37,38]
- The COG conducted a randomized trial in children with JMML that compared a standard-intensity preparative regimen (busulfan/cyclophosphamide/melphalan) with a reduced-intensity regimen (busulfan/fludarabine).[39]
- The trial closed to enrollment early when an interim analysis revealed a higher frequency of relapse/disease persistence (7 of 9 patients) in children who received the reduced-intensity regimen than in children who received the standard-intensity regimen (1 of 6 patients).
- The COG conducted a randomized trial in children with JMML that compared a standard-intensity preparative regimen (busulfan/cyclophosphamide/melphalan) with a reduced-intensity regimen (busulfan/fludarabine).[39]
Disease recurrence is the primary cause of treatment failure for children with JMML after HSCT and occurs in 30% to 40% of cases.[25,32,33] While the role of donor lymphocyte infusions is uncertain,[40] reports indicate that approximately 50% of patients with relapsed JMML can be successfully treated with a second HSCT.[41]
Treatment Options Under Clinical Evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- COG-ADVL1521 (NCT03190915) (Trametinib in Treating Patients With Relapsed or Refractory JMML): This trial is evaluating the activity of trametinib (inhibitor of MEK1/2, which is downstream of RAS/MAPK signaling) in pediatric patients with relapsed or refractory JMML. The rationale for studying this agent is based on the finding that nearly all genetic mutations found in JMML lead to aberrant RAS pathway signaling. Eligible patients are those who have relapsed or have persistent disease after intravenous chemotherapy (such as fludarabine or cytarabine) and/or HSCT, but not after low-dose oral chemotherapy (such as mercaptopurine). The primary objective is to determine the response rate of trametinib administered orally once daily in 28-day cycles.
References:
- Passmore SJ, Chessells JM, Kempski H, et al.: Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol 121 (5): 758-67, 2003.
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Chan RJ, Cooper T, Kratz CP, et al.: Juvenile myelomonocytic leukemia: a report from the 2nd International JMML Symposium. Leuk Res 33 (3): 355-62, 2009.
- Loh ML: Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol 152 (6): 677-87, 2011.
- Bresolin S, Zecca M, Flotho C, et al.: Gene expression-based classification as an independent predictor of clinical outcome in juvenile myelomonocytic leukemia. J Clin Oncol 28 (11): 1919-27, 2010.
- Olk-Batz C, Poetsch AR, Nöllke P, et al.: Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia with poor outcome. Blood 117 (18): 4871-80, 2011.
- Stiller CA, Chessells JM, Fitchett M: Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer 70 (5): 969-72, 1994.
- Choong K, Freedman MH, Chitayat D, et al.: Juvenile myelomonocytic leukemia and Noonan syndrome. J Pediatr Hematol Oncol 21 (6): 523-7, 1999 Nov-Dec.
- Tartaglia M, Niemeyer CM, Fragale A, et al.: Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34 (2): 148-50, 2003.
- Kratz CP, Niemeyer CM, Castleberry RP, et al.: The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 106 (6): 2183-5, 2005.
- Strullu M, Caye A, Lachenaud J, et al.: Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet 51 (10): 689-97, 2014.
- Loh ML, Sakai DS, Flotho C, et al.: Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 114 (9): 1859-63, 2009.
- Muramatsu H, Makishima H, Jankowska AM, et al.: Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood 115 (10): 1969-75, 2010.
- Niemeyer CM, Kang MW, Shin DH, et al.: Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet 42 (9): 794-800, 2010.
- Pérez B, Mechinaud F, Galambrun C, et al.: Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet 47 (10): 686-91, 2010.
- Hecht A, Meyer JA, Behnert A, et al.: Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 107 (1): 178-186, 2022.
- Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 (11): 1334-40, 2015.
- Stieglitz E, Taylor-Weiner AN, Chang TY, et al.: The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47 (11): 1326-33, 2015.
- Murakami N, Okuno Y, Yoshida K, et al.: Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 131 (14): 1576-1586, 2018.
- Sakaguchi H, Okuno Y, Muramatsu H, et al.: Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet 45 (8): 937-41, 2013.
- Stieglitz E, Mazor T, Olshen AB, et al.: Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat Commun 8 (1): 2127, 2017.
- Helsmoortel HH, Bresolin S, Lammens T, et al.: LIN28B overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood 127 (9): 1163-72, 2016.
- Freedman MH, Estrov Z, Chan HS: Juvenile chronic myelogenous leukemia. Am J Pediatr Hematol Oncol 10 (3): 261-7, 1988 Fall.
- Locatelli F, Nöllke P, Zecca M, et al.: Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood 105 (1): 410-9, 2005.
- Bergstraesser E, Hasle H, Rogge T, et al.: Non-hematopoietic stem cell transplantation treatment of juvenile myelomonocytic leukemia: a retrospective analysis and definition of response criteria. Pediatr Blood Cancer 49 (5): 629-33, 2007.
- Castleberry RP, Emanuel PD, Zuckerman KS, et al.: A pilot study of isotretinoin in the treatment of juvenile chronic myelogenous leukemia. N Engl J Med 331 (25): 1680-4, 1994.
- Woods WG, Barnard DR, Alonzo TA, et al.: Prospective study of 90 children requiring treatment for juvenile myelomonocytic leukemia or myelodysplastic syndrome: a report from the Children's Cancer Group. J Clin Oncol 20 (2): 434-40, 2002.
- Loh ML: Childhood myelodysplastic syndrome: focus on the approach to diagnosis and treatment of juvenile myelomonocytic leukemia. Hematology Am Soc Hematol Educ Program 2010: 357-62, 2010.
- Hasle H: Myelodysplastic and myeloproliferative disorders in children. Curr Opin Pediatr 19 (1): 1-8, 2007.
- Stieglitz E, Ward AF, Gerbing RB, et al.: Phase II/III trial of a pre-transplant farnesyl transferase inhibitor in juvenile myelomonocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (4): 629-36, 2015.
- Smith FO, King R, Nelson G, et al.: Unrelated donor bone marrow transplantation for children with juvenile myelomonocytic leukaemia. Br J Haematol 116 (3): 716-24, 2002.
- Yusuf U, Frangoul HA, Gooley TA, et al.: Allogeneic bone marrow transplantation in children with myelodysplastic syndrome or juvenile myelomonocytic leukemia: the Seattle experience. Bone Marrow Transplant 33 (8): 805-14, 2004.
- Baker D, Cole C, Price J, et al.: Allogeneic bone marrow transplantation in juvenile myelomonocytic leukemia without total body irradiation. J Pediatr Hematol Oncol 26 (3): 200-3, 2004.
- Locatelli F, Niemeyer CM: How I treat juvenile myelomonocytic leukemia. Blood 125 (7): 1083-90, 2015.
- Locatelli F, Crotta A, Ruggeri A, et al.: Analysis of risk factors influencing outcomes after cord blood transplantation in children with juvenile myelomonocytic leukemia: a EUROCORD, EBMT, EWOG-MDS, CIBMTR study. Blood 122 (12): 2135-41, 2013.
- Yabe M, Sako M, Yabe H, et al.: A conditioning regimen of busulfan, fludarabine, and melphalan for allogeneic stem cell transplantation in children with juvenile myelomonocytic leukemia. Pediatr Transplant 12 (8): 862-7, 2008.
- Koyama M, Nakano T, Takeshita Y, et al.: Successful treatment of JMML with related bone marrow transplantation after reduced-intensity conditioning. Bone Marrow Transplant 36 (5): 453-4; author reply 454, 2005.
- Dvorak CC, Satwani P, Stieglitz E, et al.: Disease burden and conditioning regimens in ASCT1221, a randomized phase II trial in children with juvenile myelomonocytic leukemia: A Children's Oncology Group study. Pediatr Blood Cancer 65 (7): e27034, 2018.
- Yoshimi A, Bader P, Matthes-Martin S, et al.: Donor leukocyte infusion after hematopoietic stem cell transplantation in patients with juvenile myelomonocytic leukemia. Leukemia 19 (6): 971-7, 2005.
- Yoshimi A, Mohamed M, Bierings M, et al.: Second allogeneic hematopoietic stem cell transplantation (HSCT) results in outcome similar to that of first HSCT for patients with juvenile myelomonocytic leukemia. Leukemia 21 (3): 556-60, 2007.
Chronic Myelogenous Leukemia (CML)
Incidence
Chronic myelogenous leukemia (CML) accounts for less than 5% of all childhood leukemia, and in the pediatric age range, occurs most commonly in older adolescents.[1]
Molecular Abnormality
The cytogenetic abnormality most characteristic of CML is the Philadelphia chromosome (Ph), which represents a translocation of chromosomes 9 and 22 (t(9;22)) resulting in a BCR::ABL1 protein fusion.[2]
Clinical Presentation
CML is characterized by a marked leukocytosis and is often associated with thrombocytosis, sometimes with abnormal platelet function. Bone marrow aspiration or biopsy reveals hypercellularity with relatively normal granulocytic maturation and no significant increase in leukemic blasts. Although reduced leukocyte alkaline phosphatase activity is seen in CML, this is not a specific finding.
CML has the following three clinical phases:
- Chronic phase. Chronic phase, which lasts for approximately 3 years if untreated, usually presents with symptoms secondary to hyperleukocytosis such as weakness, fever, night sweats, bone pain, respiratory distress, priapism, left upper quadrant pain (splenomegaly), and, rarely, hearing loss and visual disturbances.
- Accelerated phase. The accelerated phase is characterized by progressive splenomegaly, thrombocytopenia, and increased percentage of peripheral and bone marrow blasts, along with accumulation of karyotypic abnormalities in addition to the Ph chromosome.
- Blast crisis phase. Blast crisis is notable for the bone marrow, showing greater than 20% blasts or chloromatous lesions and a clinical picture that is indistinguishable from acute leukemia. Approximately two-thirds of blast crisis is myeloid, and the remainder is lymphoid, usually of B lineage. Patients in blast crisis will die within a few months.[3]
Treatment of CML: Historical Perspective
Before the tyrosine kinase inhibitor (TKI) era, allogeneic hematopoietic stem cell transplantation (HSCT) was the primary treatment for children with CML. Published reports from this period described survival rates of 70% to 80% when an HLA–matched-family donor (MFD) was used in the treatment of children in early chronic phase, with lower survival rates when HLA–matched-unrelated donors were used.[4,5,6]
Relapse rates were low (less than 20%) when transplant was performed in chronic phase.[4,5] The primary cause of death was treatment-related mortality, which was increased with HLA–matched-unrelated donors compared with HLA-MFDs in most reports.[4,5] High-resolution DNA matching for HLA alleles appeared to reduce rates of treatment-related mortality, leading to improved outcome for HSCT using unrelated donors.[7]
Compared with transplantation in chronic phase, transplantation in accelerated phase or blast crisis and in second-chronic phase resulted in significantly reduced survival.[4,5,6] The use of T-lymphocyte depletion to avoid graft-versus-host disease resulted in a higher relapse rate and decreased overall survival (OS),[8] supporting the contribution of a graft-versus-leukemia effect to favorable outcome after allogeneic HSCT.
The introduction of the TKI imatinib as a therapeutic drug targeted at inhibiting the BCR::ABL1 fusion kinase revolutionized the treatment of patients with CML, for both children and adults.[9] As most data on the use of TKIs for CML is from adult clinical trials, the adult experience is initially described, followed by a description of the more limited experience in children.
Treatment of Adult CML With TKIs
Imatinib is a potent inhibitor of the ABL1 tyrosine kinase, platelet-derived growth factor (PDGF) receptors (alpha and beta), and KIT. Imatinib treatment achieves clinical, cytogenetic, and molecular remissions (as defined by the absence of BCR::ABL1 fusion transcripts) in a high proportion of CML patients treated in chronic phase.[10]
Evidence (imatinib for adults):
- Imatinib replaced the use of recombinant interferon alfa in the initial treatment of CML on the basis of the results of a large phase III trial that compared imatinib with interferon plus cytarabine (IRIS).[11,12]
- Patients who received imatinib had higher complete cytogenetic response rates (76% vs. 14% at 18 months).[11] The rate of treatment failure diminished over time, from 3.3% and 7.5% in the first and second years of imatinib treatment, respectively, to less than 1% by the fifth year of treatment.[12]
- After censoring for patients who died from causes unrelated to CML or transplantation, the overall estimated survival rate for patients randomly assigned to imatinib was 95% at 60 months.[12]
Guidelines for imatinib treatment have been developed for adults with CML on the basis of patient response to treatment, including the timing of achieving complete hematologic response, complete cytogenetic response, and major molecular response (defined as attainment of a 3-log reduction in BCR::ABL1 gene fusion/control gene ratio).[13,14,15,16]
Poor adherence is a major reason for loss of complete cytogenetic response and imatinib failure for adult patients with CML on long-term therapy.[17] The identification of BCR::ABL1 kinase domain mutations at the time of failure or of suboptimal response to imatinib treatment also has clinical implications,[18] because there are alternative BCR::ABL1 kinase inhibitors (e.g., dasatinib and nilotinib) that maintain their activity against some (but not all) mutations that confer resistance to imatinib.[13,19,20]
Two TKIs, dasatinib and nilotinib, have been shown to be effective in patients who have an inadequate response to imatinib, although not in patients with the T315I mutation. Both dasatinib and nilotinib have also received regulatory approval for the treatment of newly diagnosed chronic-phase CML in adults, on the basis of the following studies:
- Dasatinib. Dasatinib was approved on the basis of a phase III trial that compared dasatinib (100 mg daily) with imatinib (400 mg daily).[21] There was no significant difference in progression-free survival (PFS) or OS. However, after 12 months of treatment, dasatinib was associated with a higher rate of complete cytogenetic response (83% vs. 72%, P = .001) and major molecular response (46% vs. 28%, P < .0001). Responses were achieved in a shorter time with dasatinib (P < .0001).
- Nilotinib. Nilotinib (at a dose of either 300 mg or 400 mg twice daily) was compared with imatinib (400 mg daily) in a phase III trial.[22] At 12 months, the rates of complete cytogenetic response were significantly higher for nilotinib (80% for the 300-mg dose and 78% for the 400-mg dose) than were the rates for imatinib (65%) (P < .001 for both comparisons). Also, nilotinib was associated with higher rates of major molecular response (44% for the 300-mg dose and 43% for the 400-mg dose compared with 22% for imatinib, P < .001 for both comparisons). The 300-mg twice-daily dose of nilotinib was associated with a more favorable safety profile compared with the 400-mg dose.
Because of the superiority over imatinib in terms of complete cytogenetic response rate and major molecular response rate, both dasatinib and nilotinib are extensively used as first-line therapy in adults with CML. However, despite more rapid responses with dasatinib and nilotinib than with imatinib when used as frontline therapy, PFS and OS appear to be similar for all three agents.[23,24] Additional follow-up will be required to better define the impact of these agents on long-term PFS and OS.
Bosutinib is another TKI that targets the BCR::ABL1 gene fusion and has been approved by the U.S. Food and Drug Administration (FDA) for the treatment of all phases of CML in adults who show intolerance to or whose disease shows resistance to previous therapy with another TKI. Bosutinib has not been studied in the pediatric population.
Ponatinib is a BCR::ABL1 protein fusion inhibitor that is effective against the T315I mutation.[25] Ponatinib induced objective responses in approximately 70% of heavily pretreated adults with chronic-phase CML, with responses observed regardless of the baseline BCR::ABL1 kinase domain mutation.[26] Development of ponatinib has been complicated by the high rate of vascular occlusion observed in patients receiving the agent, with arterial and venous thrombosis and occlusions (including myocardial infarction and stroke) occurring in more than 20% of treated patients.[27] Ponatinib has not been prospectively studied in the pediatric population.
For adult patients with CML who proceed to allogeneic HSCT, there is no evidence that pretransplant imatinib adversely impacts outcome.
Evidence (imatinib followed by HSCT in adults):
- A retrospective study that compared 145 patients who received imatinib before transplant with a historical cohort of 231 patients showed no difference in early hepatic toxic effects or engraftment delay.[28]
- In addition, OS, disease-free survival, relapse, and nonrelapse mortality were similar between the two cohorts.
- The only factor associated with poor outcome in the cohort that received imatinib was a poor initial response to imatinib.
- Further evidence for a lack of effect of pretransplant imatinib on posttransplant outcomes was supplied by a report from the Center for International Blood and Marrow Transplant Research; this report compared outcomes of 181 pediatric and adult subjects with CML in first chronic phase treated with imatinib before HSCT with that of 657 subjects who did not receive imatinib before HSCT.[29]
- Among the patients in first chronic phase, imatinib therapy before HSCT was associated with better OS.
- A third report of imatinib followed by allogeneic HSCT supports the efficacy of this transplantation strategy in patients with imatinib failure in first chronic phase.[13]
- The 3-year OS rate was 94% for this group (n = 37), with approximately 90% of patients achieving a complete molecular remission after HSCT.
For adult patients treated with a TKI alone (without HSCT), the optimal duration of therapy remains unknown, and most patients continue TKI treatment indefinitely. Studies are ongoing to determine the safety of discontinuing TKI therapy and the criteria most prognostic of sustained remissions after ending TKI therapy.
Evidence (length of imatinib therapy in adults):
- In an attempt to determine the length of treatment, a prospective study reported on 69 adults treated with imatinib for more than 2 years who had been in a cytogenetic major response for more than 2 years. The patients were monitored monthly and restarted on imatinib if there was evidence of molecular relapse.[30]
- Of this group, 61% of patients experienced disease relapse, with about 38% still in cytogenetic major response at 24 months.
- All of the patients who had disease recurrence responded again to the reinitiation of imatinib.
- Another study reported on 40 patients with chronic-phase CML who discontinued treatment with imatinib after at least 2 years of sustained undetectable minimal residual disease (MRD) by polymerase chain reaction (PCR).[31]
- At 24 months, the probability of sustained molecular remission for patients no longer receiving imatinib was 47.1%.
- Most relapses occurred within 4 months of stopping treatment with imatinib, and no relapses beyond 27 months were observed.
- All patients with molecular relapse demonstrated a favorable response when imatinib was restarted; with a median follow-up of 42 months, no patients had progressive disease or developed the BCR::ABL1 gene fusion.
Additional research is required before cessation of imatinib or other BCR::ABL1 targeted therapy for selected patients with CML in molecular remission can be recommended as a standard clinical practice.
Treatment of Childhood CML
Treatment options for children with CML may include the following:
- TKI therapy, such as imatinib.
Imatinib has shown a high level of activity in children with CML that is comparable with the activity observed in adults.[32,33,34,35,36]
Evidence (imatinib in children):
- In a prospective trial, 44 pediatric patients with newly diagnosed CML were treated with imatinib (260 mg/day).[36]
- The PFS rate at 36 months was 98%.
- A complete hematologic response was achieved in 98% of the patients.
- The rate of complete cytogenetic response was 61% and the rate of major molecular response was 31% at 12 months, similar to the rates seen in adult chronic-phase CML patients treated with imatinib.
As a result of this high level of activity, it is common to initiate imatinib treatment in children with CML rather than proceeding immediately to allogeneic stem cell transplantation.[37] The pharmacokinetics of imatinib in children appears consistent with previous results in adults.[38]
Doses of imatinib used in phase II trials for children with CML have ranged from 260 mg/m2 to 340 mg/m2, which provide comparable drug exposures as the adult flat-doses of 400 mg to 600 mg.[34,35,36]
Evidence (imatinib dose in children):
- In an Italian study of 47 pediatric patients with chronic-phase CML who were treated with 340 mg/m2 per day of imatinib, complete cytogenetic response was achieved in 91.5% of patients at a median time of 6 months, and the rate of major molecular response at 12 months was 66.6%.[36]
Thus, it appears that starting with the higher dose of 340 mg/m2 has superior efficacy and is typically tolerable, with dose adjustment as needed for toxicity.[35,36]
- Early molecular responses in children have been associated with improved PFS, similar to early molecular response data in adults. Early molecular response was defined as children who had a BCR::ABL1/ABL ratio of ≤10% (using a PCR-based MRD measurement) 3 months after starting imatinib.[39]
The monitoring guidelines described above for adults with CML are reasonable to use in children.
Imatinib is generally well tolerated in children, with adverse effects generally being mild to moderate and reversible with treatment discontinuation or dose reduction.[34,35] Growth retardation occurs in most prepubertal children receiving imatinib.[40] Children receiving imatinib and experiencing growth impairment may show some catch-up growth during their pubertal growth spurts, but they are at risk of having lower-than-expected adult height, as most patients do not achieve midparental height.[40,41]
There are fewer published data regarding the efficacy and toxicities of the two other TKIs approved by the FDA for use in children with CML—dasatinib and nilotinib.
Evidence (dasatinib in children):
- A phase I trial of dasatinib in children showed that drug disposition, tolerability, and efficacy of this agent was similar to that observed in adults.[42,43]
- A phase II trial of dasatinib, which included 84 children with newly diagnosed CML in chronic phase, utilized a dose of 60 mg/m2 (tablets) or 72 mg/m2 (oral solution) given to patients once daily.[44]
- Complete cytogenetic response and major molecular response (≥3-log reduction or ≤0.1% on the International Scale) were achieved in 92% and 52% of patients, respectively, after 12 months of therapy, with a 4-year PFS rate of 93%.
- Dasatinib was well tolerated, with very few grade 3 or grade 4 adverse events. No pleural or pericardial effusions or pulmonary complications were observed.
Evidence (nilotinib in children):
- The approval of nilotinib by the FDA in March 2018 for the treatment of children with CML was based on two sponsored trials.[45,46]
An initial study (NCT01077544 [CAMN107A2120]) of 11 patients evaluated pharmacokinetic, safety, and preliminary efficacy data; a second study (NCT01844765 [CAMN107A2203; AAML1321]) of 58 patients evaluated efficacy and safety. Data from both studies were combined for a pooled-data analysis of 69 patients, which included 25 patients with newly diagnosed CML and 44 patients with resistant or intolerant CML. Both studies utilized a dose of 230 mg/m2 given twice daily (rounded to the nearest 50 mg; maximum dose, 400 mg).[46]
- In the phase II trial, 64% of patients with newly diagnosed CML achieved a major molecular response at 1 year.
- The tolerability of nilotinib in children was similar to that observed in adults. Primary side effects affecting more than 30% of children included headache, fever, and hyperbilirubinemia.
- Prolongation of QTc interval (defined in this trial as an increase of >30 msec over baseline) is a recognized side effect of nilotinib, and it was observed in 25% of children in these trials. The investigators recommend obtaining an electrocardiogram at baseline, 1 week, periodically afterward, and after dose adjustments.
A safe pediatric dose has not yet been established for other TKIs (e.g., bosutinib and ponatinib).
Discontinuation of TKI therapy
Discontinuation of TKI treatment is an accepted strategy for adults with CML who meet strict criteria related to their duration of treatment and to their response to treatment. Guidelines for discontinuation of TKIs have been developed by both the European LeukemiaNet (ELN) and the U.S. National Comprehensive Cancer Network (NCCN).[47,48] Key elements for both guidelines include the following:
- TKI therapy for a minimum duration of 4 to 5 years for ELN and 3 years for NCCN.
- A minimum duration of deep molecular response (DMR or MR4) (BCR::ABL1 protein transcript level ≤0.01% International Scale [IS]) of 2 years for both ELN and NCCN.
These guidelines specify close monitoring of BCR::ABL1 transcript levels after TKI discontinuation. Loss of major molecular response (MMR or MR3) (BCR::ABL1 transcript level ≤0.1% IS) is generally used as the trigger for re-initiation of TKI therapy.
Loss of MMR is most likely to occur within the first 6 months of TKI discontinuation. Loss of MMR occurs much less frequently more than 1 year after TKI discontinuation. A meta-analysis included 3,105 adult patients who initiated a first attempt at TKI discontinuation. The study found that the probability of molecular recurrence was 35% after 0 to 6 months, 8% after 6 to 12 months, 3% after 12 to 18 months, and 3% after 18 to 24 months.[49] These results indicated that approximately 50% of adult patients maintained their molecular responses 2 years after TKI discontinuation. Relapses can occur when TKIs have been discontinued for more than 2 years, but these recurrences appear to be infrequent (<2%). Unfavorable outcomes were uncommon when relapses occurred. In addition, 90% of patients re-acquired deep molecular remission after TKI re-initiation.
There is limited data regarding TKI discontinuation in children with CML. This limited experience is explained, in part, by the low incidence of CML in children. In addition, only a minority of children with CML who are treated with TKIs meet the criteria for TKI discontinuation. For example, among patients enrolled on the International Chronic Myeloid Leukemia Pediatric Study (I-CML-Ped [NCT01281735]), only 9% of children with CML who were treated with TKIs met the criteria for TKI discontinuation.[50] Other reports have also supported this trend.[51,52] Although the small number of children studied is a limitation, it appears that the outcome for TKI discontinuation in children with CML is similar to that of adults. Two of the larger pediatric studies that discuss this topic are summarized below:
- The Japan Pediatric Leukemia and Lymphoma Study Group (JPSLG) reported on 22 children with CML who met their criteria for TKI discontinuation, which was similar to the NCCN's TKI discontinuation criteria.[52] The median age at CML diagnosis was 9 years, and the median age at TKI discontinuation was 16 years. The median duration of TKI therapy exceeded 8 years, and the median duration of MR4 before TKI discontinuation exceeded 4 years. Eleven of 22 children experienced loss of MMR at a median of 90 days after TKI discontinuation. All of these children subsequently regained MR4 after TKI resumption. The treatment-free remission rate at 12 months was 50%, and no relapses were observed beyond 4 months of TKI discontinuation.
TKI withdrawal syndrome is observed in approximately 20% to 30% of adults when TKI therapy is discontinued.[53] The syndrome includes musculoskeletal pain that typically develops within 2 months of TKI discontinuation and continues for several months. The JPLSG study did not observe musculoskeletal pain in children after TKI discontinuation.
- The International Registry of Childhood Chronic Myeloid Leukemia reported on 18 patients with CML who were younger than 18 years at diagnosis. These patients discontinued imatinib after meeting the criteria for TKI discontinuation (i.e., in chronic phase with a sustained DMR to imatinib [MR4; BCR::ABL1 transcript level ≤0.01% IS]) for at least 2 years.[50]
Among the 18 children who stopped taking imatinib, 9 (50%) eventually resumed treatment.[50] Seven of these nine patients experienced loss of MMR (BCR::ABL1 transcript level ≤0.1% IS). Six of the seven patients regained MR4 within a median of approximately 5 months after TKI re-initiation. The remaining patient achieved MMR after TKI re-initiation. Two additional patients who had a one-log increase in BCR::ABL1 transcript levels, but did not meet the criteria for loss of MMR, were restarted on imatinib by their physicians. For the other nine patients who remained in treatment-free remission, the median follow-up period after imatinib discontinuation was 50 months. TKI withdrawal syndrome was not reported in any patients discontinuing imatinib.
Treatment Options under Clinical Evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- AAML18P1 (NCT03817398) (Stopping TKIs in Affecting Treatment-Free Remission in Patients With Chronic-Phase CML): This COG study is a nonrandomized, prospective, longitudinal study enrolling children who have achieved a deep remission (BCR::ABL1 transcript level of <0.01% by real-time quantitative-polymerase chain reaction of peripheral blood [>MR4, i.e., >4-log reduction]) for at least 2 years after receiving the same TKI for at least 3 years. This trial is examining the incidence of molecular recurrence after discontinuing TKI therapy and the ability of patients to re-enter remission if recurrence occurs.
Treatment of Recurrent or Refractory Childhood CML
Treatment options for children with recurrent or refractory CML may include the following:
- Alternative kinase inhibitors such as dasatinib or nilotinib.
- Allogeneic HSCT.
In children who develop a hematologic or cytogenetic relapse during treatment with imatinib or who have an inadequate initial response to imatinib, determination of BCR::ABL1 kinase domain mutation status should be considered to help guide subsequent therapy. Depending on the patient's mutation status, alternative kinase inhibitors such as dasatinib or nilotinib can be considered on the basis of the adult and pediatric experience with these agents.[21,22,44,54,55,56]
Evidence (dasatinib in children with resistant or intolerant CML):
- In a study of 14 children with resistant or intolerant CML, the following results were observed:[44]
- 76% of patients were in complete cytogenetic remission, and 41% of patients had a major molecular response after 12 months of dasatinib therapy.
- The PFS rate was 78% at 48 months.
Evidence (nilotinib in children with resistant or intolerant CML):
- In a study of 44 children with CML who were resistant or intolerant to imatinib or dasatinib, the following results were observed:[45]
- 40.7% of patients achieved a major molecular response after 12 months of nilotinib therapy.
- After a median of 11.3 months, no patients had experienced disease progression.
Dasatinib and nilotinib are active against many BCR::ABL1 gene fusion mutations that confer resistance to imatinib, although the agents are ineffective in patients with the T315I mutation. In the presence of the T315I mutation, which is resistant to all FDA-approved kinase inhibitors, an allogeneic transplant should be considered. Ponatinib, the BCR::ABL1 protein fusion inhibitor that is effective against the T315I mutation in adults, has not been prospectively studied in the pediatric population.
The question of whether a pediatric patient with CML should receive an allogeneic transplant when multiple TKIs are available remains unanswered; however, reports suggest that PFS does not improve when using HSCT, compared with the sustained use of imatinib.[36] The potential advantages and disadvantages need to be discussed with the patient and family. While HSCT is currently the only known definitive curative therapy for CML, patients discontinuing treatment with TKIs after sustained molecular remissions, who remained in molecular remission, have been reported.[31]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649. Also available online. Last accessed August 8, 2022.
- Quintás-Cardama A, Cortes J: Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 113 (8): 1619-30, 2009.
- O'Dwyer ME, Mauro MJ, Kurilik G, et al.: The impact of clonal evolution on response to imatinib mesylate (STI571) in accelerated phase CML. Blood 100 (5): 1628-33, 2002.
- Millot F, Esperou H, Bordigoni P, et al.: Allogeneic bone marrow transplantation for chronic myeloid leukemia in childhood: a report from the Société Française de Greffe de Moelle et de Thérapie Cellulaire (SFGM-TC). Bone Marrow Transplant 32 (10): 993-9, 2003.
- Cwynarski K, Roberts IA, Iacobelli S, et al.: Stem cell transplantation for chronic myeloid leukemia in children. Blood 102 (4): 1224-31, 2003.
- Weisdorf DJ, Anasetti C, Antin JH, et al.: Allogeneic bone marrow transplantation for chronic myelogenous leukemia: comparative analysis of unrelated versus matched sibling donor transplantation. Blood 99 (6): 1971-7, 2002.
- Lee SJ, Klein J, Haagenson M, et al.: High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood 110 (13): 4576-83, 2007.
- Horowitz MM, Gale RP, Sondel PM, et al.: Graft-versus-leukemia reactions after bone marrow transplantation. Blood 75 (3): 555-62, 1990.
- Druker BJ: Translation of the Philadelphia chromosome into therapy for CML. Blood 112 (13): 4808-17, 2008.
- Kantarjian H, Sawyers C, Hochhaus A, et al.: Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med 346 (9): 645-52, 2002.
- O'Brien SG, Guilhot F, Larson RA, et al.: Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 348 (11): 994-1004, 2003.
- Druker BJ, Guilhot F, O'Brien SG, et al.: Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 355 (23): 2408-17, 2006.
- Saussele S, Lauseker M, Gratwohl A, et al.: Allogeneic hematopoietic stem cell transplantation (allo SCT) for chronic myeloid leukemia in the imatinib era: evaluation of its impact within a subgroup of the randomized German CML Study IV. Blood 115 (10): 1880-5, 2010.
- Hughes TP, Hochhaus A, Branford S, et al.: Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: an analysis from the International Randomized Study of Interferon and STI571 (IRIS). Blood 116 (19): 3758-65, 2010.
- Kantarjian H, Cortes J: Considerations in the management of patients with Philadelphia chromosome-positive chronic myeloid leukemia receiving tyrosine kinase inhibitor therapy. J Clin Oncol 29 (12): 1512-6, 2011.
- Bisen A, Claxton DF: Tyrosine kinase targeted treatment of chronic myelogenous leukemia and other myeloproliferative neoplasms. Adv Exp Med Biol 779: 179-96, 2013.
- Ibrahim AR, Eliasson L, Apperley JF, et al.: Poor adherence is the main reason for loss of CCyR and imatinib failure for chronic myeloid leukemia patients on long-term therapy. Blood 117 (14): 3733-6, 2011.
- Soverini S, Hochhaus A, Nicolini FE, et al.: BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood 118 (5): 1208-15, 2011.
- Hazarika M, Jiang X, Liu Q, et al.: Tasigna for chronic and accelerated phase Philadelphia chromosome--positive chronic myelogenous leukemia resistant to or intolerant of imatinib. Clin Cancer Res 14 (17): 5325-31, 2008.
- Brave M, Goodman V, Kaminskas E, et al.: Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin Cancer Res 14 (2): 352-9, 2008.
- Kantarjian H, Shah NP, Hochhaus A, et al.: Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 362 (24): 2260-70, 2010.
- Saglio G, Kim DW, Issaragrisil S, et al.: Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med 362 (24): 2251-9, 2010.
- Jabbour E, Kantarjian HM, Saglio G, et al.: Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION). Blood 123 (4): 494-500, 2014.
- Hochhaus A, Saglio G, Hughes TP, et al.: Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia 30 (5): 1044-54, 2016.
- O'Hare T, Shakespeare WC, Zhu X, et al.: AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16 (5): 401-12, 2009.
- Cortes JE, Kim DW, Pinilla-Ibarz J, et al.: A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med 369 (19): 1783-96, 2013.
- Prasad V, Mailankody S: The accelerated approval of oncologic drugs: lessons from ponatinib. JAMA 311 (4): 353-4, 2014 Jan 22-29.
- Oehler VG, Gooley T, Snyder DS, et al.: The effects of imatinib mesylate treatment before allogeneic transplantation for chronic myeloid leukemia. Blood 109 (4): 1782-9, 2007.
- Lee SJ, Kukreja M, Wang T, et al.: Impact of prior imatinib mesylate on the outcome of hematopoietic cell transplantation for chronic myeloid leukemia. Blood 112 (8): 3500-7, 2008.
- Mahon FX, Réa D, Guilhot J, et al.: Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 11 (11): 1029-35, 2010.
- Ross DM, Branford S, Seymour JF, et al.: Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood 122 (4): 515-22, 2013.
- Champagne MA, Capdeville R, Krailo M, et al.: Imatinib mesylate (STI571) for treatment of children with Philadelphia chromosome-positive leukemia: results from a Children's Oncology Group phase 1 study. Blood 104 (9): 2655-60, 2004.
- Millot F, Guilhot J, Nelken B, et al.: Imatinib mesylate is effective in children with chronic myelogenous leukemia in late chronic and advanced phase and in relapse after stem cell transplantation. Leukemia 20 (2): 187-92, 2006.
- Millot F, Baruchel A, Guilhot J, et al.: Imatinib is effective in children with previously untreated chronic myelogenous leukemia in early chronic phase: results of the French national phase IV trial. J Clin Oncol 29 (20): 2827-32, 2011.
- Champagne MA, Fu CH, Chang M, et al.: Higher dose imatinib for children with de novo chronic phase chronic myelogenous leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 57 (1): 56-62, 2011.
- Giona F, Putti MC, Micalizzi C, et al.: Long-term results of high-dose imatinib in children and adolescents with chronic myeloid leukaemia in chronic phase: the Italian experience. Br J Haematol 170 (3): 398-407, 2015.
- Andolina JR, Neudorf SM, Corey SJ: How I treat childhood CML. Blood 119 (8): 1821-30, 2012.
- Menon-Andersen D, Mondick JT, Jayaraman B, et al.: Population pharmacokinetics of imatinib mesylate and its metabolite in children and young adults. Cancer Chemother Pharmacol 63 (2): 229-38, 2009.
- Millot F, Guilhot J, Baruchel A, et al.: Impact of early molecular response in children with chronic myeloid leukemia treated in the French Glivec phase 4 study. Blood 124 (15): 2408-10, 2014.
- Shima H, Tokuyama M, Tanizawa A, et al.: Distinct impact of imatinib on growth at prepubertal and pubertal ages of children with chronic myeloid leukemia. J Pediatr 159 (4): 676-81, 2011.
- Millot F, Guilhot J, Baruchel A, et al.: Growth deceleration in children treated with imatinib for chronic myeloid leukaemia. Eur J Cancer 50 (18): 3206-11, 2014.
- Aplenc R, Blaney SM, Strauss LC, et al.: Pediatric phase I trial and pharmacokinetic study of dasatinib: a report from the children's oncology group phase I consortium. J Clin Oncol 29 (7): 839-44, 2011.
- Zwaan CM, Rizzari C, Mechinaud F, et al.: Dasatinib in children and adolescents with relapsed or refractory leukemia: results of the CA180-018 phase I dose-escalation study of the Innovative Therapies for Children with Cancer Consortium. J Clin Oncol 31 (19): 2460-8, 2013.
- Gore L, Kearns PR, de Martino ML, et al.: Dasatinib in Pediatric Patients With Chronic Myeloid Leukemia in Chronic Phase: Results From a Phase II Trial. J Clin Oncol 36 (13): 1330-1338, 2018.
- Novartis Pharmaceuticals Corporation: TASIGNA (nilotinib): Prescribing Information. East Hanover, NJ: Novartis, 2018. Available online. Last accessed April 7, 2022.
- Hijiya N, Maschan A, Rizzari C, et al.: Phase 2 study of nilotinib in pediatric patients with Philadelphia chromosome-positive chronic myeloid leukemia. Blood 134 (23): 2036-2045, 2019.
- Hochhaus A, Baccarani M, Silver RT, et al.: European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 34 (4): 966-984, 2020.
- National Comprehensive Cancer Network: NCCN Guidelines for Patients: Chronic Myeloid Leukemia, 2021. Plymouth Meeting, PA: National Comprehensive Cancer Network, 2021. Available online with free subscription. Last accessed August 29, 2022.
- Dulucq S, Astrugue C, Etienne G, et al.: Risk of molecular recurrence after tyrosine kinase inhibitor discontinuation in chronic myeloid leukaemia patients: a systematic review of literature with a meta-analysis of studies over the last ten years. Br J Haematol 189 (3): 452-468, 2020.
- Millot F, Suttorp M, Ragot S, et al.: Discontinuation of Imatinib in Children with Chronic Myeloid Leukemia: A Study from the International Registry of Childhood CML. Cancers (Basel) 13 (16): , 2021.
- de Bruijn CMA, Millot F, Suttorp M, et al.: Discontinuation of imatinib in children with chronic myeloid leukaemia in sustained deep molecular remission: results of the STOP IMAPED study. Br J Haematol 185 (4): 718-724, 2019.
- Shima H, Kada A, Tanizawa A, et al.: Discontinuation of tyrosine kinase inhibitors in pediatric chronic myeloid leukemia. Pediatr Blood Cancer 69 (8): e29699, 2022.
- Berger MG, Pereira B, Rousselot P, et al.: Longer treatment duration and history of osteoarticular symptoms predispose to tyrosine kinase inhibitor withdrawal syndrome. Br J Haematol 187 (3): 337-346, 2019.
- Hochhaus A, Baccarani M, Deininger M, et al.: Dasatinib induces durable cytogenetic responses in patients with chronic myelogenous leukemia in chronic phase with resistance or intolerance to imatinib. Leukemia 22 (6): 1200-6, 2008.
- le Coutre P, Ottmann OG, Giles F, et al.: Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood 111 (4): 1834-9, 2008.
- Kantarjian H, O'Brien S, Talpaz M, et al.: Outcome of patients with Philadelphia chromosome-positive chronic myelogenous leukemia post-imatinib mesylate failure. Cancer 109 (8): 1556-60, 2007.
Survivorship and Adverse Late Sequelae
While the issues of long-term complications of cancer and its treatment cross many disease categories, several important issues related to the treatment of myeloid malignancies are worth emphasizing. For more information, see Late Effects of Treatment for Childhood Cancer.
Selected studies of the late effects of AML therapy in adult survivors who were not treated with hematopoietic stem cell transplant (HSCT) include the following:
- Cardiac.
- The Childhood Cancer Survivor Study (CCSS) examined 272 survivors of childhood acute myeloid leukemia (AML) who did not undergo an HSCT.[1]
- This study identified second malignancies (cumulative incidence, 1.7%) and cardiac toxic effects (cumulative incidence, 4.7%) as significant long-term risks.
- Cardiomyopathy has been reported in 4.3% of survivors of AML based on Berlin-Frankfurt-Münster studies. Of these, 2.5% showed clinical symptoms.[2]
- A retrospective study of cardiac function of children treated with United Kingdom Medical Research Council–based regimens at a median of 13 months after treatment reported a mean detrimental change in left ventricular stroke volume of 8.4% compared with baseline values.[3]
- For pediatric patients, the risk of developing early toxicity was 13.7%, and the risk of developing late cardiac toxic effects (defined as 1 year after completing first-line therapy) was 17.4%. Early cardiac toxic effects was a significant predictor of late cardiac toxic effects and the development of clinical cardiomyopathy requiring long-term therapy.[4]
- Retrospective analysis of a single study suggests cardiac risk may be increased in children with Down syndrome,[5] but prospective studies are required to confirm this finding.
- The Childhood Cancer Survivor Study (CCSS) examined 272 survivors of childhood acute myeloid leukemia (AML) who did not undergo an HSCT.[1]
- Psychosocial.
- A Nordic Society for Pediatric Hematology and Oncology retrospective trial of children with AML treated with chemotherapy only at a median follow-up of 11 years, based on self-reported uses of health care services, demonstrated similar health care usage and marital status as their siblings.[6]
- A population-based study of survivors of childhood AML who had not undergone an HSCT reported equivalent rates of educational achievement, employment, and marital status compared with siblings. AML survivors were, however, significantly more likely to be receiving prescription drugs, especially for asthma, than were siblings (23% vs. 9%; P = .03). Chronic fatigue has also been demonstrated to be a significantly more likely adverse late effect in survivors of childhood AML than in survivors of other malignancies.[7]
- A CCSS report evaluated survivors of childhood AML treated between 1970 and 1999 (median age at the time of assessment, 30 to 32 years) and compared their outcomes to data from siblings. Survivors who received either intensive chemotherapy consolidation (n = 299) or underwent HSCT (n = 183) had statistically significant worse outcomes than did their siblings in somatic symptom measures (prevalence, 8.4%–12%), neurocognitive functioning (prevalence, 17.7%–25.7%), health-related quality-of-life measurements (prevalence, 8.2%–24.6%), and social attainment measures. In all measures, there was no statistically significant difference in prevalence of problems identified between the two consolidation cohorts.[8]
Renal, gastrointestinal, and hepatic late adverse effects have been reported to be rare for children undergoing chemotherapy only for treatment of AML.[9]
Selected studies of the late effects of AML therapy in adult survivors who were treated with HSCT include the following:
- In a review from one institution, the highest frequency of adverse long-term sequelae for children treated for AML included the following incidence rates: growth abnormalities (51%), neurocognitive abnormalities (30%), transfusion-acquired hepatitis (28%), infertility (25%), endocrinopathies (16%), restrictive lung disease (20%), chronic graft-versus-host disease (20%), secondary malignancies (14%), and cataracts (12%).[10]
- Most of these adverse sequelae are the consequence of myeloablative, allogeneic HSCT. Although cardiac abnormalities were reported in 8% of patients, this is an issue that may be particularly relevant with the current use of increased anthracyclines in clinical trials for children with newly diagnosed AML.
- Another study examined outcomes for children younger than 3 years with AML or acute lymphoblastic leukemia (ALL) who underwent HSCT.[11]
- The toxicities reported include growth hormone deficiency (59%), dyslipidemias (59%), hypothyroidism (35%), osteochondromas (24%), and decreased bone mineral density (24%).
- Two of the 33 patients developed secondary malignancies.
- Survivors had average intelligence but frequent attention-deficit problems and fine-movement abnormalities, compared with population controls.
- In contrast, The Bone Marrow Transplant Survivor Study compared childhood AML or ALL survivors with siblings using a self-reporting questionnaire.[12] The median follow-up was 8.4 years, and 86% of patients received total-body irradiation (TBI) as part of their preparative transplant regimen.
- Survivors of leukemia who received an HSCT had significantly higher frequencies of several adverse effects, including diabetes, hypothyroidism, osteoporosis, cataracts, osteonecrosis, exercise-induced shortness of breath, neurosensory impairments, and problems with balance, tremor, and weakness than did siblings.
- The overall assessment of health was significantly decreased in survivors compared with siblings (odds ratio, 2.2; P = .03).
- Significant differences were not observed between regimens using TBI compared with chemotherapy only, which mostly included busulfan.
- The outcomes were similar for patients with AML and ALL, suggesting that the primary cause underlying the adverse late effects was undergoing an HSCT.
- A Children's Oncology Group (COG) study using a health-related, quality-of-life comparison reported that 21% of 5-year survivors had a severe or life-threatening chronic health condition; when compared by type of treatment, this percentage was 16% for the chemotherapy-only treated group, 21% for the autologous HSCT treated group, and 33% for those who received an allogeneic HSCT.[13]
- A CCSS cohort analysis examined the long-term mortality and health statuses of 856 children (5-year survivors) previously treated for AML, with or without HSCT, between 1970 and 1999.[14]
- Cumulative rates of grades 3 to 5 chronic health conditions significantly declined among HSCT recipients between the 1970s and 1990s (from 76.1% to 43.5%; P = .04) but remained stable for chemotherapy-only recipients (from 12.2% to 27.6%; P = .06).
- There was a significant decline in cumulative all-cause late mortality over the same time-frame for HSCT recipients (from 38.9% to 8.5%; P < .0001). This was primarily a result of a reduction in relapse, whereas no significant decline in late mortality was seen in the chemotherapy-only survivors (from 6.8% to 2.6%; P = .35).
- In self-reports, health statuses among all survivors were excellent, very good, or good in 85% of HSCT recipients and in 90% of chemotherapy-only recipients. However, survivors' health statuses in both treatment groups were significantly worse than that of their siblings (HR, 3.8; 95% CI, 2.7–5.4 vs. HR, 2.6; 95% CI, 1.8–3.6, respectively).
New therapeutic approaches to reduce long-term adverse sequelae are needed, especially for reducing the late sequelae associated with myeloablative HSCT.
Important resources for details on follow-up and risks for survivors of cancer have been developed, including the COG's Long-Term Follow-Up Guidelines for Survivors of Childhood, Adolescent, and Young Adult Cancers and the National Comprehensive Cancer Network's Guidelines for Acute Myeloid Leukemia. Furthermore, having access to past medical history that can be shared with subsequent medical providers has become increasingly recognized as important for cancer survivors.
References:
- Mulrooney DA, Dover DC, Li S, et al.: Twenty years of follow-up among survivors of childhood and young adult acute myeloid leukemia: a report from the Childhood Cancer Survivor Study. Cancer 112 (9): 2071-9, 2008.
- Creutzig U, Diekamp S, Zimmermann M, et al.: Longitudinal evaluation of early and late anthracycline cardiotoxicity in children with AML. Pediatr Blood Cancer 48 (7): 651-62, 2007.
- Orgel E, Zung L, Ji L, et al.: Early cardiac outcomes following contemporary treatment for childhood acute myeloid leukemia: a North American perspective. Pediatr Blood Cancer 60 (9): 1528-33, 2013.
- Temming P, Qureshi A, Hardt J, et al.: Prevalence and predictors of anthracycline cardiotoxicity in children treated for acute myeloid leukaemia: retrospective cohort study in a single centre in the United Kingdom. Pediatr Blood Cancer 56 (4): 625-30, 2011.
- O'Brien MM, Taub JW, Chang MN, et al.: Cardiomyopathy in children with Down syndrome treated for acute myeloid leukemia: a report from the Children's Oncology Group Study POG 9421. J Clin Oncol 26 (3): 414-20, 2008.
- Molgaard-Hansen L, Glosli H, Jahnukainen K, et al.: Quality of health in survivors of childhood acute myeloid leukemia treated with chemotherapy only: a NOPHO-AML study. Pediatr Blood Cancer 57 (7): 1222-9, 2011.
- Jóhannsdóttir IM, Hjermstad MJ, Moum T, et al.: Increased prevalence of chronic fatigue among survivors of childhood cancers: a population-based study. Pediatr Blood Cancer 58 (3): 415-20, 2012.
- Stefanski KJ, Anixt JS, Goodman P, et al.: Long-Term Neurocognitive and Psychosocial Outcomes After Acute Myeloid Leukemia: A Childhood Cancer Survivor Study Report. J Natl Cancer Inst 113 (4): 481-495, 2021.
- Skou AS, Glosli H, Jahnukainen K, et al.: Renal, gastrointestinal, and hepatic late effects in survivors of childhood acute myeloid leukemia treated with chemotherapy only--a NOPHO-AML study. Pediatr Blood Cancer 61 (9): 1638-43, 2014.
- Leung W, Hudson MM, Strickland DK, et al.: Late effects of treatment in survivors of childhood acute myeloid leukemia. J Clin Oncol 18 (18): 3273-9, 2000.
- Perkins JL, Kunin-Batson AS, Youngren NM, et al.: Long-term follow-up of children who underwent hematopoeitic cell transplant (HCT) for AML or ALL at less than 3 years of age. Pediatr Blood Cancer 49 (7): 958-63, 2007.
- Baker KS, Ness KK, Weisdorf D, et al.: Late effects in survivors of acute leukemia treated with hematopoietic cell transplantation: a report from the Bone Marrow Transplant Survivor Study. Leukemia 24 (12): 2039-47, 2010.
- Schultz KA, Chen L, Chen Z, et al.: Health conditions and quality of life in survivors of childhood acute myeloid leukemia comparing post remission chemotherapy to BMT: a report from the children's oncology group. Pediatr Blood Cancer 61 (4): 729-36, 2014.
- Turcotte LM, Whitton JA, Leisenring WM, et al.: Chronic conditions, late mortality, and health status after childhood AML: a Childhood Cancer Survivor Study report. Blood 141 (1): 90-101, 2023.
Changes to This Summary (06 / 14 / 2023)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
General Information About Childhood Acute Myeloid Leukemia (AML)
Added hypodiploidy as a genetic abnormality associated with an unfavorable prognosis. Also added text to state that hypodiploidy is defined as a modal chromosome number of less than or equal to 45. In a retrospective cohort analysis, the International Berlin-Frankfurt-Münster AML Study Group aimed to characterize hypodiploidy in pediatric patients with AML (cited Hammer et al. as reference 111).
Treatment of Childhood AML
Added text about the results of the Children's Oncology Group (COG) AML1031 trial, which showed that sorafenib improved the event-free survival of pediatric patients with de novo AML and high-allelic ratio FLT3 internal tandem duplication mutations (cited Pollard et al. as reference 36).
Acute Promyelocytic Leukemia (APL)
Added text to state that in the COG AAML1331 trial, patients with standard-risk APL had idarubicin eliminated from the induction cycle. Mitoxantrone, high-dose cytarabine, and idarubicin were eliminated from the consolidation cycles. Mercaptopurine and methotrexate were eliminated from the maintenance cycles. Intrathecal doses of cytarabine were also eliminated.
Myelodysplastic Syndromes (MDS)
Added text to state that in one retrospective analysis, only the revised International Prognostic Scoring System (R-IPSS) very poor–risk subgroup, defined as having complex cytogenetics, was found to have a significant adverse prognostic impact on overall survival and relapse risk after transplant (cited Yamamoto et al. as reference 34).
Added text about the R-IPSS prognostic groups and associated cytogenetic abnormalities.
Chronic Myelogenous Leukemia (CML)
Added Discontinuation of TKI therapy as a new subsection.
Survivorship and Adverse Late Sequelae
Added text about the results of a Childhood Cancer Survivor Study cohort analysis that examined the long-term mortality and health statuses of 856 children previously treated for AML, with or without hematopoietic stem cell transplantation, between 1970 and 1999 (cited Turcotte et al. as reference 14).
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood acute myeloid leukemia and other myeloid malignancies. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Childhood Acute Myeloid Leukemia/Other Myeloid Malignancies Treatment are:
- William L. Carroll, MD (Laura and Isaac Perlmutter Cancer Center at NYU Langone)
- Alan Scott Gamis, MD, MPH (Children's Mercy Hospital)
- Karen J. Marcus, MD, FACR (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Jessica Pollard, MD (Dana-Farber/Boston Children's Cancer and Blood Disorders Center)
- Michael A. Pulsipher, MD (Children's Hospital Los Angeles)
- Rachel E. Rau, MD (Texas Medical Center)
- Lewis B. Silverman, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Malcolm A. Smith, MD, PhD (National Cancer Institute)
- Sarah K. Tasian, MD (Children's Hospital of Philadelphia)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Acute Myeloid Leukemia/Other Myeloid Malignancies Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/leukemia/hp/child-aml-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389454]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2023-06-14
Topic Contents
- General Information About Childhood Acute Myeloid Leukemia (AML)
- Classification of Pediatric Myeloid Malignancies
- Treatment Option Overview for Childhood AML
- Special Considerations for the Treatment of Children With Cancer
- Treatment of Childhood AML
- Acute Promyelocytic Leukemia (APL)
- Myeloid Proliferations Associated With Down Syndrome
- Myelodysplastic Syndromes (MDS)
- Therapy-Related AML and Therapy-Related Myelodysplastic Syndromes
- Juvenile Myelomonocytic Leukemia (JMML)
- Chronic Myelogenous Leukemia (CML)
- Survivorship and Adverse Late Sequelae
- Changes to This Summary (06 / 14 / 2023)
- About This PDQ Summary
This information does not replace the advice of a doctor. Healthwise, Incorporated, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the Terms of Use. Learn how we develop our content.