Genetics of Endocrine and Neuroendocrine Neoplasias (PDQ®): Genetics - Health Professional Information [NCI]
Multiple Endocrine Neoplasia Type 1
Clinical Description
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant syndrome, with an estimated prevalence of about 1 in 30,000 individuals.[1] The major endocrine features of MEN1 include the following:
- Parathyroid tumors and primary hyperparathyroidism (PHPT).
- Duodenopancreatic neuroendocrine tumors (NETs).
- Pituitary tumors.
A clinical diagnosis of MEN1 may be made when an individual has two of the three major endocrine tumors listed above, especially if he/she was diagnosed with these tumors at a young age. Alternatively, familial MEN1 may be defined as having at least one MEN1 case in the family plus at least one first-degree relative (FDR) with one of these three tumors, or two FDRs with a germline pathogenic variant.[2,3,4,5]
Initial clinical presentation of symptoms typically occurs between the ages of 20 and 30 years. However, in many cases, an MEN1 diagnosis may not be confirmed for many years after initial symptoms occur. The age-related penetrance of MEN1 is 45% to 73% by age 30 years, 82% by age 50 years, and 96% by age 70 years.[2,5,6,7] To date, there are no well-established genotype-phenotype correlations to guide clinical management of patients with MEN1.
Parathyroid Tumors and PHPT
The most common features and often the first presenting signs of MEN1 are parathyroid tumors, which result in PHPT. These tumors occur in 80% to 100% of patients by age 50 years.[8,9] MEN1-associated parathyroid tumors are typically multiglandular and hyperplastic. This differs from sporadic parathyroid tumors, which often present with a solitary adenoma.[10] The mean age of PHPT onset is 20 to 25 years in individuals with MEN1. In contrast, PHPT onset occurs in the general population at age 50 to 59 years. When MEN1 presents in childhood, the most common presenting feature is multi-gland hyperparathyroidism.[11] Parathyroid carcinoma in MEN1 is rare but has been described.[12,13,14]
Individuals with MEN1-associated PHPT will have elevated parathyroid hormone (PTH) and calcium levels in the blood. The clinical manifestations of PHPT are mainly the result of hypercalcemia. Mild hypercalcemia may go undetected and have few or no symptoms. More severe hypercalcemia can result in the following:
- Constipation.
- Nausea and vomiting.
- Dehydration.
- Decreased appetite and abdominal pain.
- Anorexia.
- Diuresis.
- Kidney stones.
- Increased bone resorption with resultant increased risk of bone fracture.
- Lethargy.
- Depression.
- Confusion.
- Hypertension.
- Shortened QT interval.
Since MEN1-associated hypercalcemia is directly related to the presence of parathyroid tumors, surgical removal of these tumors may normalize calcium and PTH levels. This can help relieve an individual's symptoms. However, there have been high recurrence rates of parathyroid tumors after surgery in some series.[15,16,17] For more information, see the Interventions section.
Duodenopancreatic NETs
Duodenopancreatic NETs are the second most common endocrine manifestation in MEN1, occurring in 30% to 80% of patients by age 40 years.[2,8] A study has shown that the incidence may be as great as twofold higher in young patients (aged 20–40 y) with pathogenic variants in exon 2 of MEN1. These individuals are also more likely to have more aggressive disease and distant metastases.[18] Furthermore, duodenopancreatic NETs are associated with early mortality even after surgical resection.[19]
Duodenopancreatic NETs seen in MEN1 include the following:
- Gastrinomas.
- Nonfunctioning NETs.
- Insulinomas.
- Vasoactive intestinal peptide tumors (VIPomas).
- Glucagonomas.
- Somatostatinomas.
| Tumor type | Estimated Penetrance | Symptoms |
|---|---|---|
| MEN1 = multiple endocrine neoplasia type 1. | ||
| Gastrinoma | ≤70%[8,20] | Peptic ulcer disease and esophagitis |
| Diarrhea | ||
| Abdominal pain | ||
| Weight loss | ||
| Nonfunctioning | 20%–55%[8,21] | Local compressive symptoms: abdominal pain, jaundice, anorexia, weight loss |
| Insulinoma | 10%[8] | Whipple's triad: symptomatic hypoglycemia reversed by glucose administration with associated elevation of insulin, C-peptide, and proinsulin levels |
| Vasoactive intestinal peptide | 1%[8,22] | Watery diarrhea |
| Hypokalemia | ||
| Achlorhydria | ||
| Glucagonoma | 1%[8,22] | Diabetes mellitus |
| Diarrhea | ||
| Depression | ||
| Necrolytic migratory erythema | ||
| Thromboembolic disease | ||
| Somatostatinoma | <1%[22] | Diabetes mellitus |
| Diarrhea/steatorrhea | ||
| Gallbladder disease | ||
| Hypochlorhydria | ||
| Weight loss | ||
Gastrinomas represent 50% of the gastrointestinal NETs in MEN1 and are the major cause of morbidity and mortality in MEN1 patients.[2,15] Gastrinomas are usually multicentric, with small (<0.5 cm) foci throughout the duodenum.[23] Most result in peptic ulcer disease (Zollinger-Ellison syndrome), and half are malignant at the time of diagnosis.[5,15,23,24]
Originally, nonfunctioning duodenopancreatic NETs were thought to be uncommon in individuals with MEN1. However, recognition of these tumors has increased with advanced genetic testing and improved imaging techniques. For example, a prospective study showed that MEN1 pathogenic variant carriers had a nonfunctioning duodenopancreatic NET frequency of 55% by age 39 years when they underwent endoscopic ultrasonography of the pancreas.[21,25] These tumors can be metastatic. One study of 108 MEN1 pathogenic variant carriers with nonfunctioning duodenopancreatic NETs showed a positive correlation between tumor size, rate of metastasis, and death. Individuals with tumors larger than 2 cm had significantly higher rates of metastasis than those with tumors smaller than 2 cm.[26] For more information, see the Molecular Genetics of MEN1 section.
Pituitary Tumors
Approximately 15% to 50% of MEN1 patients will develop a pituitary tumor.[2,8] Two-thirds are microadenomas (<1.0 cm in diameter), and the majority are prolactin-secreting.[27] Other pituitary tumors can include somatotropinomas and corticotropinomas, or they may be nonfunctioning.
| Tumor type | Estimated Penetrance | Symptoms |
|---|---|---|
| MEN1 = multiple endocrine neoplasia type 1. | ||
| Prolactinoma | 20%[8] | Galactorrhea |
| Amenorrhea/infertility | ||
| Hypogonadism | ||
| Somatotropinoma | 10%[8] | Coarse facial features |
| Soft tissue overgrowth: enlargement of hands/feet | ||
| Hyperhidrosis | ||
| Corticotropinoma | <5%[8] | Weight gain |
| Hypertension | ||
| Flushing | ||
| Easy bruising/bleeding | ||
| Hyperglycemia | ||
Other MEN1-Associated Tumors
Other manifestations of MEN1 include carcinoids of the foregut (5%–10% of MEN1 patients). These are typically bronchial or thymic and are sometimes gastric. Skin lesions are also common and can include facial angiofibromas (up to 80% of MEN1 patients) and collagenomas (~75% of MEN1 patients).[28] Lipomas (~30% of MEN1 patients) and adrenal cortical lesions (up to 50% of MEN1 patients),[29] including cortical adenomas, diffuse or nodular hyperplasia, or rarely, carcinoma are also common.[30,31,32] The following manifestations have also been reported:[33,34,35]
- Thyroid adenoma.
- Pheochromocytoma.
- Spinal ependymoma.
- Meningioma.
- Leiomyoma (e.g., esophageal, lung, and uterine).
Making the Diagnosis of MEN1
MEN1 is often difficult to diagnose in the absence of a significant family history or a positive genetic test for a pathogenic variant in the MEN1 gene. One study of 560 individuals with MEN1 showed a significant delay between the time of the first presenting symptom and the diagnosis of MEN1.[36] This time lapse is likely because some presenting symptoms of MEN1-associated tumors, such as amenorrhea, peptic ulcers, hypoglycemia, and nephrolithiasis, are not specific to MEN1.
Furthermore, identification of an MEN1-associated tumor is not sufficient to make the clinical diagnosis of MEN1 and may not trigger a referral to an endocrinologist. The median time between the first presenting symptom and diagnosis of MEN1 ranges from 7.6 years to 12 years.[6,31] Genetic testing alleviates some of this delay. Several studies have shown statistically significant differences in the age at MEN1 diagnosis between probands and their family members. In one study, clinically symptomatic probands were diagnosed with MEN1 at a mean age of 47.5 years (standard deviation [SD] +/- 13.5 y), while family members were diagnosed at a mean age of 38.5 years (SD +/- 15.4 y; P < .001).[36] In another study of 154 individuals with MEN1, probands were diagnosed at a mean age of 39.5 years (range: 18–74 y), compared with a mean age of 27 years (range: 14–56 y; P < .05) in family members diagnosed by predictive genetic testing.[37] Nonetheless, the lag time between the diagnosis of MEN1 in an index case and the diagnosis of MEN1 in family members can be significant, leading to increased morbidity and mortality.[38] This was demonstrated in a Dutch MEN1 Study Group analysis, which showed that 10% to 38% of non-index cases already had an MEN1-related manifestation at diagnosis; 4% of these individuals died of an MEN1-related cause that developed during or before the lag time. In family members, the majority of the morbidity related to lag time was due to metastatic duodenopancreatic NETs, pituitary macroadenomas, and multiple MEN1 manifestations.[38] Early intervention is particularly critical as it relates to mortality from duodenopancreatic NETs. A study showed that for every year older at time of surgery, the odds of metastasis increased by 6%.[19] These findings underscore the importance of increased awareness of the signs and symptoms of MEN1-related tumors and the constellation of findings necessary to suspect the diagnosis. It also highlights the importance of genetic counseling and testing and communication among family members once a diagnosis of MEN1 is made.[39,40] Figure 1 illustrates some of the challenges in identifying MEN1 in a family.
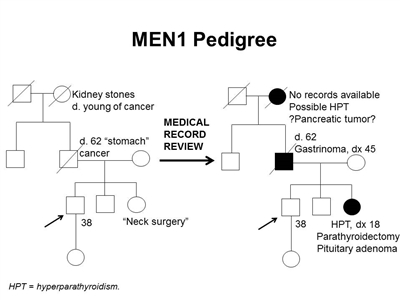
Figure 1. MEN1 pedigree. MEN1 can be very difficult to identify in a pedigree. The pedigree on the left was constructed based on self-report, and the pedigree on the right depicts the same family following a review of available medical records. This pedigree shows some of the features of a family with an MEN1 pathogenic variant across four generations, including affected family members with hyperparathyroidism, a pituitary adenoma, gastrinoma, and a suspected pancreatic tumor. The tumors in MEN1 typically occur at an earlier age than their sporadic counterparts. MEN1 families may exhibit some or all of these features. As an autosomal dominant syndrome, transmission can occur through maternal or paternal lineages, as depicted in the figure.
Since many of the tumors in MEN1 are underdiagnosed or misdiagnosed, identifying an MEN1 gene pathogenic variant in the proband early in the disease process can allow for early detection and treatment of tumors and earlier identification of at-risk family members. Many studies have been performed to determine the prevalence of MEN1 gene pathogenic variants among patients with apparently sporadic MEN1-related tumors.[8] For example, approximately one-third of patients with Zollinger-Ellison syndrome will carry an MEN1 pathogenic variant.[41,42] In individuals with apparently isolated PHPT or pituitary adenomas, the pathogenic variant prevalence is lower, on the order of 2% to 5%,[27,43,44] but the prevalence is higher in individuals diagnosed with these tumors before age 30 years. Some authors suggest referral for genetics consultation and/or genetic testing for pathogenic variants in MEN1 if one of the following conditions is present:[8,45,46,47]
- Gastrinoma at any age in the individual or an FDR.
- Multifocal duodenopancreatic NETs at any age.
- PHPT before age 30 or 40 years.
- Multiglandular parathyroid adenomas/hyperplasia or recurrent PHPT.
- Presence of one of the three main MEN1 tumors plus one of the less common tumors/findings.
- Presence of two or more features (e.g., adrenal adenomas and carcinoid tumor).
- Combination of at least two of the following in one individual: parathyroid adenoma; thymic, bronchial, or foregut carcinoid tumor; duodenopancreatic NET; pituitary tumor; adrenal tumor.
- Parathyroid adenoma and a family history of hyperparathyroidism, pituitary adenoma, duodenopancreatic NET, or foregut carcinoid tumor.
- Multiple primary duodenopancreatic NETs in the same person.
Molecular Genetics of MEN1
The MEN1 gene is located on chromosome 11q13 and encodes the protein menin.[3,48,49] Over 1,300 pathogenic variants have been identified in the MEN1 gene to date, and these are scattered across the entire coding region.[50,51] Most (~65%) of these are nonsense or frameshift variants. The remainder are missense variants (20%) (which lead to the expression of an altered protein), splice-site variants (9%), or partial- or whole-gene deletions (1%–4%).[52] In MEN1, phenotypic variation is common within a family and between unrelated individuals. Data suggested that anticipation may also occur in families with MEN1 pathogenic variants.[8,53,54,55] One large study demonstrated that pituitary, adrenal, and thymic NETs had the highest rates of heritability.[56]
Genetic Testing and Differential Diagnosis for MEN1
Genetic testing for MEN1 pathogenic variants is recommended for individuals meeting clinical diagnostic criteria and may be considered in individuals with less common MEN1-associated tumors. For more information, see the Making the diagnosis of MEN1 section. For individuals meeting diagnostic criteria, the pathogenic variant detection rate is approximately 75% to 90%.[53,57] Still, germline pathogenic variant yield ranged from 16% to 38% for apparently sporadic cases of parathyroid (15.8%), pancreatic islet (25.0%), or pituitary (37.5%) tumors. Genetic testing may be warranted in these individuals because a diagnosis of MEN1 would prompt screening for other MEN1-related tumors.[58] Laboratories that offer MEN1 testing primarily use DNA sequencing. Several laboratories offer additional analyses for MEN1 partial- or whole-gene deletion and/or duplication. However, these types of variants are rare. Deletion/duplication testing is often reserved for individuals who have very high clinical suspicions for MEN1 but a detectable pathogenic variant was not found by direct DNA sequencing. Evolving studies continue to reveal causative pathogenic variants in MEN1.[52]
A multigene panel that includes MEN1 and other genes associated with an increased risk of endocrine tumors may also be used. Such genetic testing can be used to distinguish between MEN1 and other forms of hereditary hyperparathyroidism, such as familial isolated hyperparathyroidism (FIHP), hyperparathyroidism–jaw tumor syndrome (HPT-JT), and familial hypocalciuric hypercalcemia (FHH). The hyperparathyroidism in FHH is not primary hyperparathyroidism, which is seen in MEN1, HPT-JT and FIHP. HPT-JT, which is caused by germline pathogenic variants in the CDC73 gene, is associated with PHPT, ossifying lesions of the maxilla and mandible, and renal lesions, usually bilateral renal cysts, hamartomas, and in some cases, Wilms tumor.[59,60] Unlike MEN1, HPT-JT is associated with an increased risk of parathyroid carcinoma.[61] FIHP, as its name suggests, is characterized by isolated PHPT with no additional endocrine features; in some families, FIHP is the initial diagnosis of what later develops into MEN1, HPT-JT, or FHH.[62,63,64] Approximately 20% of families with a clinical diagnosis of FIHP carry germline MEN1 pathogenic variants.[63,65,66] Pathogenic variants in the calcium-sensing receptor (CASR) gene cause FHH, which can closely mimic the hyperparathyroidism seen in MEN1.
Genetic diagnosis will help guide management for patients with early-onset hyperparathyroidism. This is especially crucial, since many of the above conditions have different management guidelines that correspond with their features. For example, distinguishing between MEN1 and FHH can be critical for a patient's disease management. Removing the parathyroid glands in FHH does not correct the hyperparathyroidism that is seen in patients with MEN1. This could result in an unnecessary surgery that would not relieve the patient's symptoms. In addition, HPT-JT is unique because it increases parathyroid carcinoma risk. Hence, individuals with this syndrome have different management guidelines than individuals with other forms of hereditary hyperparathyroidism.[67,68] For more information on MEN1 clinical features and other forms of hyperparathyroidism, see Table 3.
| Condition | Gene(s) | Major Clinical Features |
|---|---|---|
| FHH = familial hypocalciuric hypercalcemia; FIHP = familial isolated hyperparathyroidism; HPT-JT = hyperparathyroidism–jaw tumor syndrome; MEN1 = multiple endocrine neoplasia type 1 (gene is italicized); NETs = neuroendocrine tumors; PHPT = primary hyperparathyroidism. | ||
| MEN1 | MEN1 | PHPT, pituitary adenomas, duodenopancreatic NETs[8,10,69] |
| FIHP | MEN1,CDC73 | PHPT[62,63,64,65,66] |
| HPT-JT | CDC73 | PHPT; osteomas of maxilla and mandible; renal cysts or hamartomas; and rarely, Wilms tumor and parathyroid carcinoma[59,60,61] |
| FHH | CASR(type 1),GNA11(type 2),AP2S1(type 3) | Hyperparathyroidism (not primary)[67,70,71,72] |
Surveillance
Screening and surveillance for MEN1 may include a combination of biochemical tests and imaging techniques.
Traditionally, magnetic resonance imaging (MRI) was used for surveillance and staging. However, ongoing studies have evaluated the role of MRI in functional imaging, including gallium Ga 68-DOTATATE (68Ga-DOTATATE) positron emission tomography (PET)–computed tomography (CT) scanning. A multicenter retrospective study examined 108 MEN1 patients undergoing PET-CT for screening, staging, restaging, or targeted radiotherapy selection. This study demonstrated that PET-CT has the potential to increase diagnostic sensitivity when searching for MEN1-associated NETs.[73] In 51% of cases, PET-CT provided superior lesion detection when compared with conventional imaging techniques. However, the retrospective nature of the study makes it impossible to discern how much selection bias may have impacted the study's findings. Consequently, PET-CT's exact role in detecting metastasis remains unclear. PET-CT's potentially improved diagnostic sensitivity must also be weighed against its increased levels of radiation exposure, which are higher than that of other imaging modalities. Radiation exposure is particularly relevant for MEN1 patients, who are prone to developing aggressive malignancies. The issue is even more nuanced in young patients who require lifelong screening to detect aggressive MEN1-associated malignancies early.
A study analyzed thoracic screening techniques in 50 patients with MEN1. It found that when patients with MEN1 underwent functional imaging with fluorine F 18-fludeoxyglucose (18F-FDG) PET-CT screening, they had a similar number of lung nodules as individuals in the general population. However, when lesions in MEN1 patients were FDG-avid, they were more likely to progress during the follow-up period. Therefore, further observation and follow up of FDG-avid lesions may be warranted in patients with MEN1.[74] While lung-specific imaging is not routinely performed in MEN1 patients, the lungs are visualized during MRI surveillance of thymic lesions. When pulmonary lesions are identified, it is important to recognize that lung NETs may grow slowly and may have a good prognosis. Referral to a multidisciplinary team may be beneficial for selective resection of lung NETs.[75]
Recommendations for MEN1 surveillance are summarized in Table 4.[4,8]
| Biochemical Test or Procedure | Condition Screened For | Age Screening Initiated (y) | Frequency |
|---|---|---|---|
| CT = computed tomography; MEN1 = multiple endocrine neoplasia type 1; MRI = magnetic resonance imaging; NETs = neuroendocrine tumors; PHPT = primary hyperparathyroidism; PTH = parathyroid hormone. | |||
| a Adapted from Brandi et al.[4]and Thakker et al.[8] | |||
| b The recommendations for abdominal imaging differ between two published guidelines for the diagnosis and management of MEN1.[4,8]There is weak evidence at this time to support annual imaging before age 10 years. Imaging before age 10 years does identify disease in a high proportion of patients, but it is not clear whether this impacts prognosis.[21,76] | |||
| c The age to initiate screening and the screening frequency for pituitary tumors may be debatable because the clinical significance of small, nonfunctional tumors is unclear;[77]further study may be warranted. | |||
| d Adapted from Niederle et al.[5] | |||
| e Adapted from Shirali et al.[78]The 2012 guidelines recommend chest MRI every 1-2 years.[8] | |||
| Serum prolactin and/or insulin-like growth factor 1 | Pituitary tumors | 5 | Every 1 y |
| Fasting total serum calcium and/or ionized calcium and PTH | Parathyroid tumors and PHPT | 8 | Every 1 y |
| Fasting serum gastrin | Duodenopancreatic gastrinoma | 20 | Every 1 y |
| Chromogranin A, pancreatic polypeptide, glucagon, and vasointestinal polypeptided | Duodenopancreatic NETs | 10–16 | Up to every 3 years (consider every 3 years if asymptomatic; consider shorter screening intervals depending on the clinical scenario) |
| Fasting glucose and insulin | Insulinoma | 5 | Every 1 y |
| Brain MRIc | Pituitary tumors | 5 | Every 3–5 y based on biochemical results |
| Chest MRIe | Thymic and bronchial NETs | <20 | About every 3 years. Consider more frequent screening for men, smokers, or individuals with a positive family history. Baseline chest MRI is done prior to parathyroidectomy |
| Abdominal CT or MRIb[4] | Duodenopancreatic NETs | 20 | Every 3–5 y based on biochemical results |
| Abdominal CT, MRI, or endoscopic ultrasonographyb[8] | Duodenopancreatic NETs | <10 | Every 1 y |
Interventions
Surgical management of MEN1 is complex and controversial, given the multifocal and multiglandular nature of the disease. Patients with MEN1 have a high risk of tumor recurrence, even after surgery. Additionally, these patients may have an increased risk of developing venous thromboembolisms.[79] Clinicians should be aware of this risk, particularly in the perioperative period. It is critical to establish an MEN1 diagnosis before making surgical decisions, in order to prevent unnecessary and/or inappropriate surgeries. Furthermore, it is recommended that individuals with MEN1 use a surgeon who has experience treating this disease.
Treatment for parathyroid tumors
Once evidence of parathyroid disease is established biochemically, surgical removal of the hyperfunctional parathyroid tissue is recommended to achieve eucalcemia and euparathyroidism. However, the timing and the amount of parathyroid and thymus gland tissue that is removed during surgery remains controversial.[40] For patients with primary hyperparathyroidism who are at risk for MEN1, preoperative detection of a pathogenic variant helps guide the extent of their initial operations, can increase the likelihood of successful initial surgeries, and lower the likelihood of recurrent disease.[68] Furthermore, knowledge of an MEN1 pathogenic variant can help guide surgical decision making and avoid the use of a single-gland surgical approach. Studies have shown that concomitant bilateral cervical thymectomy decreases the rate of parathyroid tumor recurrence and suggests that the thymus can be removed during the patient's initial operation.[80]
Some groups reserve surgical intervention for symptomatic patients, with continued annual biochemical screening for those without objective signs of disease. Subtotal parathyroidectomy (removal of 3–3.5 glands) is commonly suggested as the initial surgical treatment when a provider decides to proceed with surgery.[68] If 3.5 or more glands are removed during surgery, the rate of persistent disease is only 5% to 6%. Removing fewer than 3.5 glands decreases the durability of eucalcemia. Studies suggest that preoperative imaging (to determine which glands are hyperfunctional) is not reliable enough to justify unilateral exploration, with 86% of patients having enlarged contralateral parathyroid glands.[81] Fifty percent of the patients who had imaging to direct resection had the largest parathyroid gland identified intraoperatively on the contralateral side of greatest uptake.[81] Insufficient resection fails to accomplish the desired eucalcemia.[15,16,17,68]
Total parathyroidectomy with autotransplantation of parathyroid tissue to a distant site, such as the forearm, is a less commonly recommended option. Likelihood of cancer recurrence is lowered with total parathyroidectomy. However, this procedure also renders the patient aparathyroid for a period of time while the autotransplanted tissue becomes functional. This can cause a permanent PTH deficiency (no detectable PTH in the body).[80,82] Benefits of this approach include the following: 1) it is easier remove/debulk recurrent disease from the forearm than it is to remove/debulk recurrent disease from the neck, and 2) differential lateralization with arm blood draws. Hypocalcemia management involves taking numerous oral medications, including calcitrol and calcium replacement, and these daily requirements can be a major burden for patients. Recovery was less likely for patients who were aparathyroid for 6 months after total parathyroidectomy.[83] In select high-risk patients, like in those who had reoperative parathyroidectomy or cervical surgery, cryopreservation can be beneficial, but it is recommended that providers use this method sparingly.
Treatment for duodenopancreatic NETs
The timing and extent of surgery for duodenopancreatic NETs are controversial and depend on many factors, including severity of symptoms, extent of disease, functional component, location and necessity of simple enucleation, subtotal or total pancreatectomy, and pancreaticoduodenectomy (Whipple procedure). Surgical enucleation has been associated with higher recurrence compared with distal pancreatectomy, and a decreased rate of endocrine insufficiency compared with a Whipple procedure.[84] Tumor size has been suggested to advocate for surgical resection on the basis of the increased propensity for risk of metastases or recurrence with increased tumor diameter.[5,85,86] Unfortunately, there is no specific tumor marker or combination of tumor markers that are predictive of disease-specific mortality.[87] Long-acting somatostatin analogs may have a role in early-stage MEN1 duodenopancreatic NETs.[88] Initial study results of pharmacological therapy suggest that the treatment is safe and that long-term suppression of tumor and hormonal activity can be seen in up to 10% of patients and stability of hormone hyperfunction in 80% of patients.[88] The primary goal of surgery is to improve long-term survival by reducing symptoms associated with hormone excess and lowering the risk of distant metastasis.[24] Surgery is commonly performed for most functional tumors and for nonfunctioning NETs when the tumor exceeds 2 to 3 cm because the likelihood of distant metastases is high.[86,89,90,91] Structural imaging modalities alone are suboptimal for predicting the malignant potential of duodenopancreatic NETs. However, a study found that screening MEN1 patients with 18F-FDG PET-CT identified those NETs with an increased malignant potential; the FDG avidity correlated with a Ki-67 index.[92] Tumor size does seem to influence patient survival, with patients with smaller tumors having increased survival after resection.[93] While more-extensive surgical approaches (e.g., pancreatoduodenectomy) have been associated with higher cure rates and improved overall survival,[94,95,96] they also have higher rates of postoperative complications and long-term morbidity.[97] Therefore, the risks and benefits should be carefully considered, and surgical decisions should be made on a case-by-case basis. With regard to open or laparoscopic approaches, in selected patients, pancreatic laparoscopic surgery appears to be safe and associated with a shorter length of stay and fewer complications.[98]
Individuals with MEN1 who are diagnosed with NETs often have multiple tumors of various types throughout the pancreas and duodenum, some of which can be identified using magnetic resonance imaging or computed tomography (CT). Combining functional tracer accumulation with anatomic imaging improves tumor localization. 68Ga-DOTATATE PET-CT demonstrates excellent sensitivity in mapping duodenopancreatic NET disease. This modality may guide the initial workup and appears to be superior to standard somatostatin octreotide, especially for lesions smaller than 10 mm.[99,100] Many tumors are too small to be detected using standard imaging techniques, and intra-arterial secretin stimulation testing and/or intraoperative ultrasonography may also be useful.[101,102] Preoperative assessment using a combination of various biochemical and imaging modalities, intraoperative assessment of tumor burden, and resolution of hormonal hyper-secretion are critical and, in some series, have been associated with higher cure rates and longer disease-free intervals.[101,102,103,104]
In the current era of effective treatment for hyperfunctional hormone excess states, most MEN1-related deaths are due to the malignant nature of duodenopancreatic NETs. A less common but important risk of death is from malignant thymic carcinoid tumors. Indicators of a poor MEN1 prognosis include elevated fasting serum gastrin, the presence of functional hormonal syndromes, liver or distant metastases, aggressive duodenopancreatic NET growth, large duodenopancreatic NET size, or the need for multiple parathyroidectomies. The most common cause of non-MEN1–related death in this patient cohort is from cardiovascular disease.[105]
Other duodenopancreatic NETs
Glucagonomas, VIPomas, and somatostatinomas are rare but often have higher rates of malignancy than other duodenopancreatic NETs.[22] These are often treated with aggressive surgery.[106]
Insulinomas
Medical management of insulinoma using diet and medication is often unsuccessful; the mainstay of treatment for this tumor is surgical resection.[8] Insulinomas in MEN1 patients can be located throughout the pancreas, with a preponderance found in the distal gland,[107,108,109] and have a higher rate of metastasis than sporadic insulinoma.[106] Surgery can range from enucleation of single or multiple large tumors to partial pancreatic resection, or both,[108] to subtotal or total pancreatectomy.[107,108] More-extensive surgical approaches are associated with a lower rate of recurrence [94,95,108,110] but a higher rate of postoperative morbidity. Because insulinoma often occurs in conjunction with nonfunctioning pancreatic tumors, the selective intra-arterial calcium-injection test (SAS test) may be necessary to determine the source of insulin excess.[111] Intraoperative monitoring of insulin/glucose can help determine whether insulin-secreting tumors have been successfully excised.[102,112]
Gastrinomas
Most MEN1-associated gastrinomas originate in the duodenum. These tumors are typically multifocal and cause hyper-secretion of gastrin, with resultant peptic ulcer disease (Zollinger-Ellison syndrome).[113] The multifocal nature makes complete surgical resection difficult. It is critical to manage symptoms before considering any type of surgical intervention.[114] Historically, some groups have recommended close observation of individuals with smaller tumors (<2.0 cm on imaging) who have relief of symptoms using medications (e.g., proton pump inhibitors or histamine-2 agonists);[115] however, this approach may not be optimal for all patients.
Several published series have shown a positive correlation between primary tumor size and rate of distant metastasis. One retrospective study showed that 61% of patients with tumors larger than 3 cm had liver metastases.[24] In another series, 40% of patients with tumors larger than 3 cm had liver metastases.[116] In contrast, both of these series showed significantly lower rates of liver metastases in individuals with tumors smaller than 3 cm (32% and 4.8%, respectively). On the basis of these and other data, many groups recommend surgery in individuals with nonmetastatic gastrinoma who have tumors larger than 2 cm.[8,96]
The type of surgery for gastrinoma depends on many factors. A Whipple procedure is typically discouraged as an initial surgery, given the high postoperative morbidity and long-term complications, such as diabetes mellitus and malabsorption. Less extensive operations have been described with varying results. At a minimum, duodenectomy with intraoperative palpation and/or ultrasonography to locate and excise duodenal tumors and peri-pancreatic lymph node dissection are performed.[101,117] Because most patients with gastrinoma will have concomitant NETs throughout the pancreas, some of which may be nonfunctional, some groups recommend resection of the distal pancreas and enucleation of tumors in the pancreatic head in addition to duodenal tumor excision.[101,117,118]
Nonfunctioning NETs
Approximately 50% of individuals with MEN1 will develop nonfunctioning NETs.[21,26] These are often identified incidentally during assessment and exploration for functioning tumors. As with gastrinomas, the metastatic rate is correlated with larger tumor size.[26,86] Tumors smaller than 1.5 cm are not likely to have lymph node metastases,[119] although the presence of metastatic disease has been associated with earlier age at death than in those without duodenopancreatic NETs.[9,26]
Pituitary tumors
Medical therapy to suppress hypersecretion is often the first line of therapy for MEN1-associated pituitary tumors. In one series of 136 patients, medical therapy was successful in approximately one-half of patients with secreting tumors (49 of 116, 42%), and successful suppression was correlated with smaller tumor size.[120] Surgery is often necessary for patients who are resistant to this treatment. Radiation therapy is reserved for patients for whom complete surgical resection was not rendered.[8,121]
References:
- Agarwal SK, Ozawa A, Mateo CM, et al.: The MEN1 gene and pituitary tumours. Horm Res 71 (Suppl 2): 131-8, 2009.
- Trump D, Farren B, Wooding C, et al.: Clinical studies of multiple endocrine neoplasia type 1 (MEN1) QJM 89 (9): 653-69, 1996.
- Chandrasekharappa SC, Guru SC, Manickam P, et al.: Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 276 (5311): 404-7, 1997.
- Brandi ML, Gagel RF, Angeli A, et al.: Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86 (12): 5658-71, 2001.
- Niederle B, Selberherr A, Bartsch DK, et al.: Multiple Endocrine Neoplasia Type 1 and the Pancreas: Diagnosis and Treatment of Functioning and Non-Functioning Pancreatic and Duodenal Neuroendocrine Neoplasia within the MEN1 Syndrome - An International Consensus Statement. Neuroendocrinology 111 (7): 609-630, 2021.
- Carty SE, Helm AK, Amico JA, et al.: The variable penetrance and spectrum of manifestations of multiple endocrine neoplasia type 1. Surgery 124 (6): 1106-13; discussion 1113-4, 1998.
- Goudet P, Dalac A, Le Bras M, et al.: MEN1 disease occurring before 21 years old: a 160-patient cohort study from the Groupe d'étude des Tumeurs Endocrines. J Clin Endocrinol Metab 100 (4): 1568-77, 2015.
- Thakker RV, Newey PJ, Walls GV, et al.: Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 97 (9): 2990-3011, 2012.
- Goudet P, Murat A, Binquet C, et al.: Risk factors and causes of death in MEN1 disease. A GTE (Groupe d'Etude des Tumeurs Endocrines) cohort study among 758 patients. World J Surg 34 (2): 249-55, 2010.
- Chandrasekharappa SC, Teh BT: Clinical and molecular aspects of multiple endocrine neoplasia type 1. Front Horm Res 28: 50-80, 2001.
- Shariq OA, Lines KE, English KA, et al.: Multiple endocrine neoplasia type 1 in children and adolescents: Clinical features and treatment outcomes. Surgery 171 (1): 77-87, 2022.
- del Pozo C, García-Pascual L, Balsells M, et al.: Parathyroid carcinoma in multiple endocrine neoplasia type 1. Case report and review of the literature. Hormones (Athens) 10 (4): 326-31, 2011 Oct-Dec.
- Christakis I, Busaidy NL, Cote GJ, et al.: Parathyroid carcinoma and atypical parathyroid neoplasms in MEN1 patients; A clinico-pathologic challenge. The MD Anderson case series and review of the literature. Int J Surg 31: 10-6, 2016.
- Singh Ospina N, Sebo TJ, Thompson GB, et al.: Prevalence of parathyroid carcinoma in 348 patients with multiple endocrine neoplasia type 1 - case report and review of the literature. Clin Endocrinol (Oxf) 84 (2): 244-249, 2016.
- Norton JA, Venzon DJ, Berna MJ, et al.: Prospective study of surgery for primary hyperparathyroidism (HPT) in multiple endocrine neoplasia-type 1 and Zollinger-Ellison syndrome: long-term outcome of a more virulent form of HPT. Ann Surg 247 (3): 501-10, 2008.
- Hellman P, Skogseid B, Oberg K, et al.: Primary and reoperative parathyroid operations in hyperparathyroidism of multiple endocrine neoplasia type 1. Surgery 124 (6): 993-9, 1998.
- Schreinemakers JM, Pieterman CR, Scholten A, et al.: The optimal surgical treatment for primary hyperparathyroidism in MEN1 patients: a systematic review. World J Surg 35 (9): 1993-2005, 2011.
- Christakis I, Qiu W, Hyde SM, et al.: Genotype-phenotype pancreatic neuroendocrine tumor relationship in multiple endocrine neoplasia type 1 patients: A 23-year experience at a single institution. Surgery 163 (1): 212-217, 2018.
- Donegan D, Singh Ospina N, Rodriguez-Gutierrez R, et al.: Long-term outcomes in patients with multiple endocrine neoplasia type 1 and pancreaticoduodenal neuroendocrine tumours. Clin Endocrinol (Oxf) 86 (2): 199-206, 2017.
- Norton JA, Krampitz G, Jensen RT: Multiple Endocrine Neoplasia: Genetics and Clinical Management. Surg Oncol Clin N Am 24 (4): 795-832, 2015.
- Thomas-Marques L, Murat A, Delemer B, et al.: Prospective endoscopic ultrasonographic evaluation of the frequency of nonfunctioning pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. Am J Gastroenterol 101 (2): 266-73, 2006.
- Lévy-Bohbot N, Merle C, Goudet P, et al.: Prevalence, characteristics and prognosis of MEN 1-associated glucagonomas, VIPomas, and somatostatinomas: study from the GTE (Groupe des Tumeurs Endocrines) registry. Gastroenterol Clin Biol 28 (11): 1075-81, 2004.
- Pipeleers-Marichal M, Somers G, Willems G, et al.: Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med 322 (11): 723-7, 1990.
- Weber HC, Venzon DJ, Lin JT, et al.: Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: a prospective long-term study. Gastroenterology 108 (6): 1637-49, 1995.
- Tonelli F, Giudici F, Fratini G, et al.: Pancreatic endocrine tumors in multiple endocrine neoplasia type 1 syndrome: review of literature. Endocr Pract 17 (Suppl 3): 33-40, 2011 Jul-Aug.
- Triponez F, Dosseh D, Goudet P, et al.: Epidemiology data on 108 MEN 1 patients from the GTE with isolated nonfunctioning tumors of the pancreas. Ann Surg 243 (2): 265-72, 2006.
- Corbetta S, Pizzocaro A, Peracchi M, et al.: Multiple endocrine neoplasia type 1 in patients with recognized pituitary tumours of different types. Clin Endocrinol (Oxf) 47 (5): 507-12, 1997.
- Darling TN, Skarulis MC, Steinberg SM, et al.: Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol 133 (7): 853-7, 1997.
- Ventura M, Melo M, Carrilho F: Outcome and long-term follow-up of adrenal lesions in multiple endocrine neoplasia type 1. Arch Endocrinol Metab 63 (5): 516-523, 2019.
- Machens A, Schaaf L, Karges W, et al.: Age-related penetrance of endocrine tumours in multiple endocrine neoplasia type 1 (MEN1): a multicentre study of 258 gene carriers. Clin Endocrinol (Oxf) 67 (4): 613-22, 2007.
- Pieterman CR, Schreinemakers JM, Koppeschaar HP, et al.: Multiple endocrine neoplasia type 1 (MEN1): its manifestations and effect of genetic screening on clinical outcome. Clin Endocrinol (Oxf) 70 (4): 575-81, 2009.
- Waldmann J, Bartsch DK, Kann PH, et al.: Adrenal involvement in multiple endocrine neoplasia type 1: results of 7 years prospective screening. Langenbecks Arch Surg 392 (4): 437-43, 2007.
- Gibril F, Schumann M, Pace A, et al.: Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore) 83 (1): 43-83, 2004.
- McKeeby JL, Li X, Zhuang Z, et al.: Multiple leiomyomas of the esophagus, lung, and uterus in multiple endocrine neoplasia type 1. Am J Pathol 159 (3): 1121-7, 2001.
- Vortmeyer AO, Lubensky IA, Skarulis M, et al.: Multiple endocrine neoplasia type 1: atypical presentation, clinical course, and genetic analysis of multiple tumors. Mod Pathol 12 (9): 919-24, 1999.
- Yamazaki M, Suzuki S, Kosugi S, et al.: Delay in the diagnosis of multiple endocrine neoplasia type 1: typical symptoms are frequently overlooked. Endocr J 59 (9): 797-807, 2012.
- Lourenço DM, Toledo RA, Coutinho FL, et al.: The impact of clinical and genetic screenings on the management of the multiple endocrine neoplasia type 1. Clinics (Sao Paulo) 62 (4): 465-76, 2007.
- van Leeuwaarde RS, van Nesselrooij BP, Hermus AR, et al.: Impact of Delay in Diagnosis in Outcomes in MEN1: Results From the Dutch MEN1 Study Group. J Clin Endocrinol Metab 101 (3): 1159-65, 2016.
- Strømsvik N, Nordin K, Berglund G, et al.: Living with multiple endocrine neoplasia type 1: decent care-insufficient medical and genetic information: a qualitative study of MEN 1 patients in a Swedish hospital. J Genet Couns 16 (1): 105-17, 2007.
- Marini F, Giusti F, Tonelli F, et al.: Management impact: effects on quality of life and prognosis in MEN1. Endocr Relat Cancer 24 (10): T227-T242, 2017.
- Roy PK, Venzon DJ, Shojamanesh H, et al.: Zollinger-Ellison syndrome. Clinical presentation in 261 patients. Medicine (Baltimore) 79 (6): 379-411, 2000.
- Bardram L, Stage JG: Frequency of endocrine disorders in patients with the Zollinger-Ellison syndrome. Scand J Gastroenterol 20 (2): 233-8, 1985.
- Uchino S, Noguchi S, Sato M, et al.: Screening of the Men1 gene and discovery of germ-line and somatic mutations in apparently sporadic parathyroid tumors. Cancer Res 60 (19): 5553-7, 2000.
- Scheithauer BW, Laws ER, Kovacs K, et al.: Pituitary adenomas of the multiple endocrine neoplasia type I syndrome. Semin Diagn Pathol 4 (3): 205-11, 1987.
- Newey PJ, Thakker RV: Role of multiple endocrine neoplasia type 1 mutational analysis in clinical practice. Endocr Pract 17 (Suppl 3): 8-17, 2011 Jul-Aug.
- Hampel H, Bennett RL, Buchanan A, et al.: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 17 (1): 70-87, 2015.
- Bashford MT, Kohlman W, Everett J, et al.: Addendum: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 21 (12): 2844, 2019.
- Larsson C, Skogseid B, Oberg K, et al.: Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 332 (6159): 85-7, 1988.
- Bassett JH, Forbes SA, Pannett AA, et al.: Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet 62 (2): 232-44, 1998.
- Lemos MC, Thakker RV: Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 29 (1): 22-32, 2008.
- Concolino P, Costella A, Capoluongo E: Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years. Cancer Genet 209 (1-2): 36-41, 2016 Jan-Feb.
- Brandi ML, Agarwal SK, Perrier ND, et al.: Multiple Endocrine Neoplasia Type 1: Latest Insights. Endocr Rev 42 (2): 133-170, 2021.
- Giraud S, Zhang CX, Serova-Sinilnikova O, et al.: Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am J Hum Genet 63 (2): 455-67, 1998.
- Wautot V, Vercherat C, Lespinasse J, et al.: Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: search for correlation between phenotype and the functional domains of the MEN1 protein. Hum Mutat 20 (1): 35-47, 2002.
- van den Broek MFM, van Nesselrooij BPM, Pieterman CRC, et al.: Clues For Genetic Anticipation In Multiple Endocrine Neoplasia Type 1. J Clin Endocrinol Metab 105 (7): , 2020.
- Thevenon J, Bourredjem A, Faivre L, et al.: Unraveling the intrafamilial correlations and heritability of tumor types in MEN1: a Groupe d'étude des Tumeurs Endocrines study. Eur J Endocrinol 173 (6): 819-26, 2015.
- Agarwal SK, Kester MB, Debelenko LV, et al.: Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet 6 (7): 1169-75, 1997.
- Klein RD, Salih S, Bessoni J, et al.: Clinical testing for multiple endocrine neoplasia type 1 in a DNA diagnostic laboratory. Genet Med 7 (2): 131-8, 2005.
- Teh BT, Farnebo F, Kristoffersson U, et al.: Autosomal dominant primary hyperparathyroidism and jaw tumor syndrome associated with renal hamartomas and cystic kidney disease: linkage to 1q21-q32 and loss of the wild type allele in renal hamartomas. J Clin Endocrinol Metab 81 (12): 4204-11, 1996.
- Carpten JD, Robbins CM, Villablanca A, et al.: HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet 32 (4): 676-80, 2002.
- Marx SJ: Multiple endocrine neoplasia type 1. In: Vogelstein B, Kinzler KW, eds.: The Genetic Basis of Human Cancer. McGraw-Hill, 1998, pp 489-506.
- Warner J, Epstein M, Sweet A, et al.: Genetic testing in familial isolated hyperparathyroidism: unexpected results and their implications. J Med Genet 41 (3): 155-60, 2004.
- Mizusawa N, Uchino S, Iwata T, et al.: Genetic analyses in patients with familial isolated hyperparathyroidism and hyperparathyroidism-jaw tumour syndrome. Clin Endocrinol (Oxf) 65 (1): 9-16, 2006.
- Cetani F, Pardi E, Borsari S, et al.: Molecular pathogenesis of primary hyperparathyroidism. J Endocrinol Invest 34 (7 Suppl): 35-9, 2011.
- Miedlich S, Lohmann T, Schneyer U, et al.: Familial isolated primary hyperparathyroidism--a multiple endocrine neoplasia type 1 variant? Eur J Endocrinol 145 (2): 155-60, 2001.
- Cetani F, Pardi E, Ambrogini E, et al.: Genetic analyses in familial isolated hyperparathyroidism: implication for clinical assessment and surgical management. Clin Endocrinol (Oxf) 64 (2): 146-52, 2006.
- Raue F, Frank-Raue K: Primary hyperparathyroidism--what the nephrologist should know--an update. Nephrol Dial Transplant 22 (3): 696-9, 2007.
- Romero Arenas MA, Morris LF, Rich TA, et al.: Preoperative multiple endocrine neoplasia type 1 diagnosis improves the surgical outcomes of pediatric patients with primary hyperparathyroidism. J Pediatr Surg 49 (4): 546-50, 2014.
- Thakker RV: Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol 386 (1-2): 2-15, 2014.
- Christensen SE, Nissen PH, Vestergaard P, et al.: Familial hypocalciuric hypercalcaemia: a review. Curr Opin Endocrinol Diabetes Obes 18 (6): 359-70, 2011.
- Nesbit MA, Hannan FM, Howles SA, et al.: Mutations affecting G-protein subunit α11 in hypercalcemia and hypocalcemia. N Engl J Med 368 (26): 2476-2486, 2013.
- Nesbit MA, Hannan FM, Howles SA, et al.: Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet 45 (1): 93-7, 2013.
- Mennetrey C, Le Bras M, Bando-Delaunay A, et al.: Value of Somatostatin Receptor PET/CT in Patients With MEN1 at Various Stages of Their Disease. J Clin Endocrinol Metab 107 (5): e2056-e2064, 2022.
- So A, Pointon O, Hodgson R, et al.: An assessment of 18 F-FDG PET/CT for thoracic screening and risk stratification of pulmonary nodules in multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf) 88 (5): 683-691, 2018.
- van den Broek MFM, de Laat JM, van Leeuwaarde RS, et al.: The Management of Neuroendocrine Tumors of the Lung in MEN1: Results From the Dutch MEN1 Study Group. J Clin Endocrinol Metab 106 (2): e1014-e1027, 2021.
- Langer P, Kann PH, Fendrich V, et al.: Prospective evaluation of imaging procedures for the detection of pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. World J Surg 28 (12): 1317-22, 2004.
- de Laat JM, Dekkers OM, Pieterman CR, et al.: Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the DutchMEN1 Study Group (DMSG). J Clin Endocrinol Metab 100 (9): 3288-96, 2015.
- Shirali AS, Pieterman CRC, Lewis MA, et al.: It's not a mystery, it's in the history: Multidisciplinary management of multiple endocrine neoplasia type 1. CA Cancer J Clin 71 (5): 369-380, 2021.
- Lee ME, Ortega-Sustache YM, Agarwal SK, et al.: Patients With MEN1 Are at an Increased Risk for Venous Thromboembolism. J Clin Endocrinol Metab 106 (2): e460-e468, 2021.
- Pieterman CR, van Hulsteijn LT, den Heijer M, et al.: Primary hyperparathyroidism in MEN1 patients: a cohort study with longterm follow-up on preferred surgical procedure and the relation with genotype. Ann Surg 255 (6): 1171-8, 2012.
- Nilubol N, Weinstein LS, Simonds WF, et al.: Limited Parathyroidectomy in Multiple Endocrine Neoplasia Type 1-Associated Primary Hyperparathyroidism: A Setup for Failure. Ann Surg Oncol 23 (2): 416-23, 2016.
- Lairmore TC, Govednik CM, Quinn CE, et al.: A randomized, prospective trial of operative treatments for hyperparathyroidism in patients with multiple endocrine neoplasia type 1. Surgery 156 (6): 1326-34; discussion 1334-5, 2014.
- Landry JP, Pieterman CRC, Clemente-Gutierrez U, et al.: Evaluation of risk factors, long-term outcomes, and immediate and delayed autotransplantation to minimize postsurgical hypoparathyroidism in multiple endocrine neoplasia type 1 (MEN1): A retrospective cohort study. Surgery 171 (5): 1240-1246, 2022.
- Ratnayake CBB, Loveday BP, Windsor JA, et al.: Patient characteristics and clinical outcomes following initial surgical intervention for MEN1 associated pancreatic neuroendocrine tumours: A systematic review and exploratory meta-analysis of the literature. Pancreatology 19 (3): 462-471, 2019.
- Kishi Y, Shimada K, Nara S, et al.: Basing treatment strategy for non-functional pancreatic neuroendocrine tumors on tumor size. Ann Surg Oncol 21 (9): 2882-8, 2014.
- Nell S, Verkooijen HM, Pieterman CRC, et al.: Management of MEN1 Related Nonfunctioning Pancreatic NETs: A Shifting Paradigm: Results From the DutchMEN1 Study Group. Ann Surg 267 (6): 1155-1160, 2018.
- Qiu W, Christakis I, Silva A, et al.: Utility of chromogranin A, pancreatic polypeptide, glucagon and gastrin in the diagnosis and follow-up of pancreatic neuroendocrine tumours in multiple endocrine neoplasia type 1 patients. Clin Endocrinol (Oxf) 85 (3): 400-7, 2016.
- Ramundo V, Del Prete M, Marotta V, et al.: Impact of long-acting octreotide in patients with early-stage MEN1-related duodeno-pancreatic neuroendocrine tumours. Clin Endocrinol (Oxf) 80 (6): 850-5, 2014.
- Triponez F, Goudet P, Dosseh D, et al.: Is surgery beneficial for MEN1 patients with small (< or = 2 cm), nonfunctioning pancreaticoduodenal endocrine tumor? An analysis of 65 patients from the GTE. World J Surg 30 (5): 654-62; discussion 663-4, 2006.
- Bettini R, Partelli S, Boninsegna L, et al.: Tumor size correlates with malignancy in nonfunctioning pancreatic endocrine tumor. Surgery 150 (1): 75-82, 2011.
- Triponez F, Sadowski SM, Pattou F, et al.: Long-term Follow-up of MEN1 Patients Who Do Not Have Initial Surgery for Small ≤2 cm Nonfunctioning Pancreatic Neuroendocrine Tumors, an AFCE and GTE Study: Association Francophone de Chirurgie Endocrinienne & Groupe d'Etude des Tumeurs Endocrines. Ann Surg 268 (1): 158-164, 2018.
- Kornaczewski Jackson ER, Pointon OP, Bohmer R, et al.: Utility of FDG-PET Imaging for Risk Stratification of Pancreatic Neuroendocrine Tumors in MEN1. J Clin Endocrinol Metab 102 (6): 1926-1933, 2017.
- Brunner SM, Weber F, Werner JM, et al.: Neuroendocrine tumors of the pancreas: a retrospective single-center analysis using the ENETS TNM-classification and immunohistochemical markers for risk stratification. BMC Surg 15: 49, 2015.
- Bartsch DK, Langer P, Wild A, et al.: Pancreaticoduodenal endocrine tumors in multiple endocrine neoplasia type 1: surgery or surveillance? Surgery 128 (6): 958-66, 2000.
- Bartsch DK, Fendrich V, Langer P, et al.: Outcome of duodenopancreatic resections in patients with multiple endocrine neoplasia type 1. Ann Surg 242 (6): 757-64, discussion 764-6, 2005.
- Norton JA, Jensen RT: Role of surgery in Zollinger-Ellison syndrome. J Am Coll Surg 205 (4 Suppl): S34-7, 2007.
- Lopez CL, Waldmann J, Fendrich V, et al.: Long-term results of surgery for pancreatic neuroendocrine neoplasms in patients with MEN1. Langenbecks Arch Surg 396 (8): 1187-96, 2011.
- Drymousis P, Raptis DA, Spalding D, et al.: Laparoscopic versus open pancreas resection for pancreatic neuroendocrine tumours: a systematic review and meta-analysis. HPB (Oxford) 16 (5): 397-406, 2014.
- Morgat C, Vélayoudom-Céphise FL, Schwartz P, et al.: Evaluation of (68)Ga-DOTA-TOC PET/CT for the detection of duodenopancreatic neuroendocrine tumors in patients with MEN1. Eur J Nucl Med Mol Imaging 43 (7): 1258-66, 2016.
- Lastoria S, Marciello F, Faggiano A, et al.: Role of (68)Ga-DOTATATE PET/CT in patients with multiple endocrine neoplasia type 1 (MEN1). Endocrine 52 (3): 488-94, 2016.
- Imamura M, Komoto I, Ota S, et al.: Biochemically curative surgery for gastrinoma in multiple endocrine neoplasia type 1 patients. World J Gastroenterol 17 (10): 1343-53, 2011.
- Tonelli F, Fratini G, Nesi G, et al.: Pancreatectomy in multiple endocrine neoplasia type 1-related gastrinomas and pancreatic endocrine neoplasias. Ann Surg 244 (1): 61-70, 2006.
- Lewis MA, Thompson GB, Young WF: Preoperative assessment of the pancreas in multiple endocrine neoplasia type 1. World J Surg 36 (6): 1375-81, 2012.
- van Asselt SJ, Brouwers AH, van Dullemen HM, et al.: EUS is superior for detection of pancreatic lesions compared with standard imaging in patients with multiple endocrine neoplasia type 1. Gastrointest Endosc 81 (1): 159-167.e2, 2015.
- Ito T, Igarashi H, Uehara H, et al.: Causes of death and prognostic factors in multiple endocrine neoplasia type 1: a prospective study: comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Medicine (Baltimore) 92 (3): 135-81, 2013.
- Akerström G, Stålberg P: Surgical management of MEN-1 and -2: state of the art. Surg Clin North Am 89 (5): 1047-68, 2009.
- O'Riordain DS, O'Brien T, van Heerden JA, et al.: Surgical management of insulinoma associated with multiple endocrine neoplasia type I. World J Surg 18 (4): 488-93; discussion 493-4, 1994 Jul-Aug.
- Crippa S, Zerbi A, Boninsegna L, et al.: Surgical management of insulinomas: short- and long-term outcomes after enucleations and pancreatic resections. Arch Surg 147 (3): 261-6, 2012.
- Sakurai A, Yamazaki M, Suzuki S, et al.: Clinical features of insulinoma in patients with multiple endocrine neoplasia type 1: analysis of the database of the MEN Consortium of Japan. Endocr J 59 (10): 859-66, 2012.
- Vezzosi D, Cardot-Bauters C, Bouscaren N, et al.: Long-term results of the surgical management of insulinoma patients with MEN1: a Groupe d'étude des Tumeurs Endocrines (GTE) retrospective study. Eur J Endocrinol 172 (3): 309-19, 2015.
- Grant CS: Insulinoma. Best Pract Res Clin Gastroenterol 19 (5): 783-98, 2005.
- Giudici F, Nesi G, Brandi ML, et al.: Surgical management of insulinomas in multiple endocrine neoplasia type 1. Pancreas 41 (4): 547-53, 2012.
- Plöckinger U: Diagnosis and Treatment of Gastrinomas in Multiple Endocrine Neoplasia Type 1 (MEN-1). Cancers (Basel) 4 (1): 39-54, 2012.
- Falconi M, Eriksson B, Kaltsas G, et al.: ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 103 (2): 153-71, 2016.
- Mignon M, Cadiot G: Diagnostic and therapeutic criteria in patients with Zollinger-Ellison syndrome and multiple endocrine neoplasia type 1. J Intern Med 243 (6): 489-94, 1998.
- Cadiot G, Vuagnat A, Doukhan I, et al.: Prognostic factors in patients with Zollinger-Ellison syndrome and multiple endocrine neoplasia type 1. Groupe d'Etude des Néoplasies Endocriniennes Multiples (GENEM and groupe de Recherche et d'Etude du Syndrome de Zollinger-Ellison (GRESZE). Gastroenterology 116 (2): 286-93, 1999.
- Dickson PV, Rich TA, Xing Y, et al.: Achieving eugastrinemia in MEN1 patients: both duodenal inspection and formal lymph node dissection are important. Surgery 150 (6): 1143-52, 2011.
- Akerström G, Stålberg P, Hellman P: Surgical management of pancreatico-duodenal tumors in multiple endocrine neoplasia syndrome type 1. Clinics (Sao Paulo) 67 (Suppl 1): 173-8, 2012.
- Zhang IY, Zhao J, Fernandez-Del Castillo C, et al.: Operative Versus Nonoperative Management of Nonfunctioning Pancreatic Neuroendocrine Tumors. J Gastrointest Surg 20 (2): 277-83, 2016.
- Vergès B, Boureille F, Goudet P, et al.: Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab 87 (2): 457-65, 2002.
- Pieterman CR, Vriens MR, Dreijerink KM, et al.: Care for patients with multiple endocrine neoplasia type 1: the current evidence base. Fam Cancer 10 (1): 157-71, 2011.
Multiple Endocrine Neoplasia Type 2
Clinical Description
The endocrine disorders observed in multiple endocrine neoplasia type 2 (MEN2) are medullary thyroid cancer (MTC); its precursor, C-cell hyperplasia (CCH) (referred to as C-cell neoplasia or C-cell carcinoma in situ in more recent publications)[1]; pheochromocytoma (PHEO); and parathyroid adenomas and/or hyperplasia. MEN2-associated MTC is often bilateral and/or multifocal and arises in the background of CCH clonal C-cell proliferation. In contrast, sporadic MTC is typically unilateral and/or unifocal. Because approximately 75% to 80% of sporadic cases also have associated CCH, this histopathologic feature cannot be used as a predictor of familial disease.[2] Metastatic spread of MTC to regional lymph nodes (i.e., perithyroidal, paratracheal, jugular chain, and upper mediastinum) or to distant sites, such as the liver, is common in patients who present with a palpable thyroid mass or diarrhea.[3,4] When thyroidectomy is performed before local nodal metastases occur, distant metastases are rare.[5] Metastatic PHEOs have not been reported in individuals with MEN2.[6] In MEN2, parathyroid abnormalities can range from benign parathyroid adenomas or multigland hyperplasia to clinically evident hyperparathyroidism with hypercalcemia and renal stones.
Historically, individuals with MEN2 were given one of the following clinical subtypes based on the presence or absence of certain endocrine tumors in the individual/family:
- MEN2A.
- Familial medullary thyroid cancer (FMTC).
- MEN2B (sometimes referred to as MEN3).
Current stratification has moved away from a solely phenotype -based classification to one that is based on genotype (i.e., the pathogenic variant) and phenotype.[7] Current classification now includes two MEN2 syndromes: MEN2A and MEN2B. The MEN2A syndrome is further classified on the basis of the presence of associated conditions. For example, classical MEN2A includes those with MTC, PHEO, and/or hyperparathyroidism. Additional categories include MEN2A with cutaneous lichen amyloidosis, MEN2A with Hirschsprung disease (HSCR), and FMTC (presence of a RETgermline pathogenic variant and MTC but no family history of PHEO or hyperparathyroidism).[1] Classifying a patient or family by MEN2 subtype is useful in determining prognosis and management.
The prevalence of MEN2 has been estimated to be approximately 1 in 35,000 individuals.[8] The vast majority of MEN2 cases are MEN2A.
MTC and CCH
MTC originates in calcitonin-producing cells (C-cells) of the thyroid gland. MTC is diagnosed when nests of C-cells extend beyond the basement membrane and infiltrate and destroy thyroid follicles. CCH is a controversial diagnosis, but most pathologists agree that it is defined as more than seven C-cells per cluster, complete follicles surrounded by C-cells, and C-cells in a distribution beyond normal anatomical location.[1,9,10,11] Individuals with RET pathogenic variants and CCH are at substantially increased risk of progressing to MTC. MTC and CCH are suspected in the presence of an elevated plasma calcitonin concentration.
A study of 10,864 patients with nodular thyroid disease found 44 (1 of every 250) cases of MTC after stimulation with calcitonin, none of which were clinically suspected. Consequently, half of these patients had no evidence of MTC on fine-needle biopsy and thus might not have undergone surgery without the positive calcitonin stimulation test.[12] CCH associated with a positive calcitonin stimulation test occurs in about 5% of the general population; therefore, the plasma calcitonin responses to stimulation do not always distinguish CCH from small MTC and cannot always distinguish between carriers and noncarriers in an MEN2 family.[13,14,15]
MTC accounts for 1% to 2% of new cases of thyroid cancer diagnosed annually in the United States.[16] Approximately 75% of thyroid cancer cases diagnosed in the United States are sporadic (i.e., they occur in the absence of a family history of either MTC or other endocrine abnormalities seen in MEN2). The peak incidence of the sporadic form is in the fifth and sixth decades of life.[3,17] A study in the United Kingdom estimated the incidence of MTC at 20 to 25 new cases per year among a population of 55 million.[18]
In the absence of a positive family history, MEN2 may be suspected when MTC occurs at an early age or is bilateral or multifocal. While small series of apparently sporadic MTC cases have suggested a higher prevalence of germline RET pathogenic variants,[19,20] larger series indicate a prevalence range of 1% to 7%.[21,22] On the basis of these data, testing for pathogenic variants in the RETgene is widely recommended for all cases of MTC.[1,23]
Level of evidence (Screening): 3
Natural history of MTC
Thyroid cancer represents approximately 2.2% of new malignancies occurring annually in the United States, with an estimated 43,720 cancer diagnoses and 2,120 cancer deaths expected in 2023.[24] Of these cancer diagnoses, 1% to 2% are MTC.[16]
MTC arises from the parafollicular calcitonin-secreting cells of the thyroid gland. MTC occurs in sporadic and familial forms and may be preceded by CCH, although CCH is a relatively common abnormality in middle-aged adults.[9,10]
Average survival for MTC is lower than that for more common thyroid cancers (e.g., 86%–89% 5-year survival for MTC compared with 94%–98% 5-year survival for papillary and follicular thyroid cancer).[16,25] Survival is correlated with stage at diagnosis, and decreased survival in MTC can be accounted for in part by a high proportion of late-stage diagnosis.[26,27,28]
In addition to early stage at diagnosis, other factors associated with improved survival in MTC include smaller tumor size, younger age at diagnosis, and diagnosis by biochemical or genetic screening (i.e., screening for calcitonin elevation, RET variants) versus symptoms.[27,29,30,31]
A Surveillance, Epidemiology, and End Results population-based study of 1,252 MTC patients found that survival varied by extent of local disease. For example, the 10-year survival rates ranged from 95.6% for those with disease confined to the thyroid gland to 40% for those with distant metastases.[29]
Hereditary MTC
While most MTC cases are sporadic, approximately 20% to 25% are hereditary caused by pathogenic variants in the RET proto-oncogene.[32] Pathogenic variants in the RET gene cause MEN2, an autosomal dominant disorder associated with a high lifetime risk of MTC. Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant endocrinopathy that is genetically and clinically distinct from MEN2; however, the similar nomenclature for MEN1 and MEN2 may cause confusion. Individuals with MEN1 do not have an increased risk of developing thyroid cancer. For more information, see the MEN1 section.
MEN2-Related PHEO
PHEOs arise from the catecholamine-producing chromaffin cells of the adrenal medulla. They are relatively rare tumors and are suspected among patients with refractory hypertension or when biochemical screening reveals elevated excretion of catecholamines and catecholamine metabolites (i.e., norepinephrine, epinephrine, metanephrine, and vanillylmandelic acid) in 24-hour urine collections or plasma. In the past, measurement of urinary catecholamines was considered the preferred biochemical screening method. However, given that catecholamines are only released intermittently and are metabolized in the adrenal medulla into metanephrine and normetanephrine, the measurement of urine or plasma fractionated metanephrines has become the gold standard.[33,34,35,36,37,38] When biochemical screening in an individual who has or is at risk of MEN2 suggests PHEO, localization studies, such as magnetic resonance imaging (MRI) or computed tomography, can be performed.[39] Confirmation of the diagnosis can be made using iodine I 131-metaiodobenzylguanidine scintigraphy or positron emission tomography imaging.[14,39,40,41]
In individuals with a personal history of PHEO, an MEN2 diagnosis is often considered in those with bilateral PHEOs, those with early age of PHEO onset (age <35 y), and those with a personal and/or family history of MTC or hyperparathyroidism. For more information, see the Familial PHEO and PGL Syndrome section. However, MEN2 is not the only genetic disorder with a predisposition to PHEOs; other disorders include neurofibromatosis type 1 (NF1), von Hippel-Lindau disease (VHL),[42] and the hereditary paraganglioma syndromes.[43] For more information on VHL, see von Hippel-Lindau Disease.
Primary Hyperparathyroidism (PHPT)
PHPT is the third most common endocrine disorder in the general population. The incidence increases with age with the vast majority of cases occurring after the sixth decade of life. Approximately 80% of cases are the result of a single adenoma.[44] PHPT can also be seen as a component tumor in several different hereditary syndromes, including the following:
Hereditary PHPT is typically multiglandular, presents earlier in life, and can have histologic evidence of both adenoma and glandular hyperplasia.
Clinical Diagnosis of MEN2 Subtypes
The diagnosis of the two MEN2 clinical subtypes relies on a combination of clinical findings, family history, and molecular genetic testing of the RET gene.
MEN2A
Classical MEN2A
MEN2A is diagnosed clinically by the occurrence of two specific endocrine tumors in addition to MTC: PHEO and/or parathyroid adenoma and/or hyperplasia in a single individual or in close relatives.[1]
The classical MEN2A subtype makes up about 60% to 90% of MEN2 cases. The MEN2A subtype was initially called Sipple syndrome.[48] Since genetic testing for RET pathogenic variants has become available, it has become apparent that about 95% of individuals with MEN2A will develop MTC.[14,49,50,51]
MTC is generally the first manifestation of MEN2A. In asymptomatic at-risk individuals, stimulation testing may reveal elevated plasma calcitonin levels and the presence of CCH or MTC.[14,50] In families with MEN2A, the biochemical manifestations of MTC generally appear between the ages of 5 years and 25 years (mean, 15 y).[14] If presymptomatic screening is not performed, MTC typically presents as a neck mass or neck pain between the ages of about age 5 years and 20 years. More than 50% of such patients have cervical lymph node metastases.[3] Diarrhea, the most frequent systemic symptom, occurs in patients with a markedly elevated plasma calcitonin level or bulky disease and/or hepatic metastases and implies a poor prognosis.[1,3,52,53] Up to 30% of patients with MTC present with diarrhea and advanced disease.[54]
MEN2-associated PHEOs are more often bilateral, multifocal, and associated with extratumoral medullary hyperplasia.[55,56,57] They also have an earlier age of onset and are less likely to be malignant than their sporadic counterparts.[55,58] MEN2-associated PHEOs usually present after MTC, typically with intractable hypertension.[59]
Unlike the PHPT seen in MEN1, hyperparathyroidism in individuals with MEN2 is typically asymptomatic or associated with only mild elevations in calcium.[54,60] A series of 56 patients with MEN2-related hyperparathyroidism has been reported by the French Calcitonin Tumors Study Group.[60] The median age at diagnosis was 38 years, documenting that this disorder is rarely the first manifestation of MEN2. This is in sharp contrast to MEN1, in which the vast majority of patients (87%–99%) initially present with primary hyperparathyroidism.[61,62,63] Parathyroid abnormalities were found concomitantly with surgery for medullary thyroid cancer in 43 patients (77%). Two-thirds of the patients were asymptomatic. Among the 53 parathyroid glands removed surgically, there were 24 single adenomas, 4 double adenomas, and 25 hyperplastic glands.
MEN2A with cutaneous lichen amyloidosis
A small number of families with MEN2A have pruritic skin lesions known as cutaneous lichen amyloidosis. This lichenoid skin lesion is located over the upper portion of the back and may appear before the onset of MTC.[64,65]
MEN2A with Hirschsprung disease (HSCR)
HSCR, a disorder of the enteric plexus of the colon that typically results in enlargement of the bowel and constipation or obstipation in neonates, occurs in a small number of individuals with MEN2A-associated RET pathogenic variants.[66] Pathogenic variants at specific cysteine residues in exon 10 (i.e., codons 609, 618, and 620) are most commonly associated with HSCR, although individuals with pathogenic variants in other exons can still be affected.[67] HSCR can occur outside of a diagnosis of MEN2A and infants with HSCR may benefit from their own genetic evaluation regardless of the likelihood of MEN2A because HSCR can present as part of other syndromes. Up to 40% of familial cases of HSCR and 3% to 7% of sporadic cases are associated with germline pathogenic variants in the RET proto-oncogene.[68,69] Certain loss-of-function RET variants have been associated with isolated HSCR,[70] indicating that not all individuals with HSCR and a germline RET variant necessarily have MEN2A.
Figure 2 depicts some of the classic manifestations of MEN2A in a family.
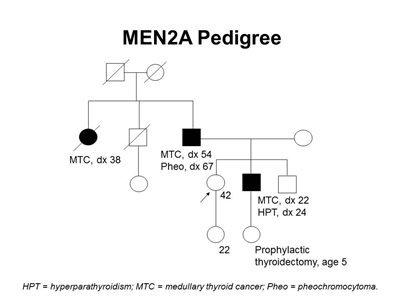
Figure 2. MEN2A pedigree. This pedigree shows some of the classic features of a family with a RET pathogenic variant across four generations, including affected family members with medullary thyroid cancer, pheochromocytoma, and hyperparathyroidism. Age at onset can vary widely, even within families. MEN2A families may exhibit some or all of these features. As an autosomal dominant syndrome, transmission can occur through maternal or paternal lineages.
Familial medullary thyroid cancer (FMTC)
Up to 50% of MEN2A cases are of the FMTC subtype, and are defined as families or single individuals with germline RET pathogenic variants and MTC alone in the absence of PHEO or parathyroid adenoma/hyperplasia.[1] This definition replaces previous classification of FMTC as a stand-alone diagnosis.[1] Previously, misclassification of families as having FMTC (because of too-small family size or later onset of other manifestations of MEN2A) could result in overlooking the risk of PHEO, a disease with significant morbidity and mortality. For this reason, FMTC is now considered a subtype of MEN2A in which there is a lack of or delay in the onset of the other (nonthyroidal) manifestations of the MEN2A syndrome.[71] Current management guidelines [1] recommend that patients thought to have pure FMTC also be screened for PHEO and hyperparathyroidism.
MEN2B
MEN2B is clinically characterized by the presence of mucosal neuromas on the lips and tongue, ganglioneuromatosis of the gastrointestinal (GI) tract, medullated corneal nerve fibers, and distinctive facies which include the following: enlarged lips, an asthenic Marfanoid body habitus, and MTC.[72,73,74,75] In cases of de novo pathogenic variants, the diagnosis of MEN2B is often delayed, after the development of MTC. The MTC is often fatal, particularly in the presence of metastatic disease, which is common at the time of diagnosis. It is important, therefore, for pediatricians to recognize the endocrine and nonendocrine clinical manifestations of the syndrome as an earlier diagnosis may result in lifesaving treatment of MTC, before metastatic spread.[76]
The MEN2B subtype makes up about 5% of MEN2 cases. The MEN2B subtype was initially called mucosal neuroma syndrome or Wagenmann-Froboese syndrome.[77] MEN2B is characterized by the early development of an aggressive form of MTC in all patients.[72] Patients with MEN2B who do not undergo thyroidectomy at an early age (at approximately age 1 y) are likely to develop metastatic MTC at an early age. Before intervention with early risk-reducing thyroidectomy, the average age at death in patients with MEN2B was 21 years. PHEOs occur in about 50% of MEN2B cases; about half are multiple and often bilateral. Clinically apparent parathyroid disease is very uncommon.[49,72] Patients with MEN2B may be identified in infancy or early childhood by a distinctive facial appearance and the presence of mucosal neuromas on the anterior dorsal surface of the tongue, palate, or pharynx. The lips become prominent over time, and submucosal nodules may be present on the vermilion border of the lips. Neuromas of the eyelids may cause thickening and eversion of the upper eyelid margins. Prominent thickened corneal nerves may be seen by slit lamp examination.
Patients with MEN2B may have diffuse ganglioneuromatosis of the gastrointestinal tract with associated symptoms that include abdominal distension, megacolon, constipation, and diarrhea.[78,79] A review of the literature reported the presence of constipation as a common symptom in 72.7% of patients with MEN2B. Additionally, gastrointestinal symptoms occurred during the first year of life in 52.3% of patients with MEN2B. Intestinal biopsy led to the diagnosis of ganglioneuromatosis in 27.3% of patients.[80]
About 75% of patients have a Marfanoid habitus, often with kyphoscoliosis or lordosis, joint laxity, and decreased subcutaneous fat. Proximal muscle wasting and weakness can also be seen.[74,75]
A retrospective review of the clinical presentation of 35 cases of MEN2B with de novo pathogenic variants treated at a single institution found that 22 cases were diagnosed because of endocrine manifestations of the syndrome.[76] The diagnosis of PHEO, a neck mass, and/or skeletal abnormalities led to the identification of MTC. The remaining 13 patients presented with a nonendocrine manifestation, including oral neuromas, corneal nerve abnormalities, persistent diarrhea, failure to thrive, or skeletal abnormalities with frequent falls. Of the entire cohort, 21 patients had one or more physician referrals for the evaluation of an MEN2B-related feature, an average of 5 years before the diagnosis of MEN2B.
It is critical for pediatricians and other providers who care for infants/children (e.g., gastroenterologists, pathologists, oral health care professionals) to maintain a high index of suspicion when evaluating patients with any of the clinical manifestations associated with MEN2B. In a child or infant, the presence of oral and ocular neuromas, GI manifestations (like severe constipation and/or the need for a rectal biopsy), and/or a tall, lanky appearance may warrant further investigation. The identification of these features may prompt an early diagnosis of MEN2B and the opportunity to prevent or cure a patient's MTC.[79,81] Some authors have recommended referral to genetic counseling for an individual with MTC or any of the following features:[72,81,82]
- Benign oral and submucosal neuromas.
- Elongated face and large lips.
- Ganglioneuromatosis.
- Inability to cry tears (biologic mechanism unknown).
Molecular Genetics of MEN2
MEN2 syndromes are the result of inherited pathogenic variants in the RET gene, located on chromosome region 10q11.2.[83,84,85] The RET gene is a proto-oncogene composed of 21 exons over 55 kilobase of genomic material.[86,87]
RET encodes a receptor tyrosine kinase with extracellular, transmembrane, and intracellular domains. Details of RET receptor and ligand interaction in this signaling pathway have been reviewed.[32,88,89] Briefly, the extracellular domain consists of a calcium-binding cadherin-like region and a cysteine-rich region that interacts with one of four ligands identified to date. These ligands, e.g., glial cell line–derived neurotrophic factor (GDNF), neurturin, persephin, and artemin, also interact with one of four coreceptors in the GDNF-family receptor–alpha family.[88] The tyrosine kinase catalytic core is located in the intracellular domain, which causes downstream signaling events through a variety of second messenger molecules.
Genetic testing for MEN2
MEN2 is a well-defined hereditary cancer syndrome. Genetic testing is an important management tool for individuals and/or family members who are at risk for MEN2. The American Thyroid Association and NCCN recommend that all patients with MTC consider genetic testing, even if they have other known risk factors that contribute to MTC.[1,23] MEN2 meets the criteria related to indications for genetic testing for cancer susceptibility outlined by the American Society of Clinical Oncology.[90] At-risk individuals are defined as first-degree relatives (parents, siblings, and children) of a person known to have MEN2. Testing can identify people with asymptomatic MEN2; these individuals can consider risk-reducing thyroidectomy and biochemical screening as preventive measures. A negative pathogenic variant analysis in at-risk relatives, however, is informative only after a disease-causing variant has been identified in an affected relative. For more information, see Cancer Genetics Risk Assessment and Counseling. Because early detection of at-risk individuals affects medical management, testing children without MEN2 symptoms can be beneficial.[91,92] For more information on clinical management of at-risk individuals, see the Genotype-Phenotype Correlations and Risk Stratification section.
Germline DNA testing for RET pathogenic variants is generally recommended to all individuals with a diagnosis of MTC, regardless of whether there is a personal or family history suggestive of MEN2.[93,94] Approximately 95% of patients with MEN2A or MEN2B will have an identifiable germline RET pathogenic variant.[95] Between 1% and 10% of individuals with apparently sporadic MTC will carry a germline RET pathogenic variant, underscoring the importance of testing all individuals diagnosed with MTC.[96,97,98]
There is no evidence for the involvement of other genetic loci, and all pathogenic variant–negative families analyzed to date have demonstrated linkage to the RET gene. For families that do not have a detectable pathogenic variant, clinical recommendations can be based on the clinical features in the affected individual and in the family.
There is considerable diversity in the techniques used and the approach to RET pathogenic variant testing among the various laboratories that perform this procedure. Methods used to detect variants in RET include polymerase chain reaction (PCR) followed by restriction enzyme digestion of PCR products, heteroduplex analysis, single-stranded conformation polymorphism analysis, denaturing high-performance liquid chromatography, and DNA sequencing.[95,99] Most testing laboratories, at a minimum, offer testing using a targeted exon approach; that is, the laboratories look for variants in the exons that are most commonly found to carry variants (exons 10, 11, 13, 14, 15 and 16). Other laboratories offer testing of all RET exons. If targeted-exon testing in a family with a high clinical suspicion for MEN2 is normal, sequencing of the remaining exons can then be performed. Differences in variant detection methods are important to consider when selecting a genetic testing laboratory and interpreting the test results. For more information on clinical validity, see Cancer Genetics Risk Assessment and Counseling.
Genotype-Phenotype Correlations and Risk Stratification
Genotype-phenotype correlations in MEN2 are well-established and have long been used to guide clinicians in making medical management recommendations. Several groups have developed pathogenic variant–stratification tables based on clinical phenotype, age of onset, and aggressiveness of MTC.[1,93,100] This classification strategy was first put forth after the Seventh International Workshop on MEN in 2001, which provided guidelines for the age of genetic testing and prophylactic thyroidectomy.[93] This stratification has been revised by the American Thyroid Association (ATA).[1,101,102] The specific pathogenic variants and their ATA classification are summarized in Table 5 below.
ATA-Highest Risk (HST) (previously labeled ATA-D) pathogenic variants are the most aggressive and carry the highest risk of developing MTC.[1] This category includes those with MEN2B and RET codon M918T pathogenic variants and is associated with the youngest age at disease onset and the highest risk of mortality. ATA-High Risk (H) (previously called ATA-C) pathogenic variants, at codons 634 and A883F, are associated with a slightly lower risk, yet the MTC in patients with these pathogenic variants is still quite aggressive and may present at an early age.[103] Former ATA-levels A and B pathogenic variants are now combined into a single group called Moderate Risk (MOD) and are associated with a lower risk of aggressive MTC relative to the risk seen in carriers of ATA-HST and ATA-H pathogenic variants.[102] Results from a study of 387 RET pathogenic variant carriers with MTC have suggested that ATA-MOD variants may be associated with MTC as aggressive as seen in individuals with ATA-H variants but present at a later age.[104] The risk of MTC is still substantially elevated over the general population risk and consideration of risk-reducing thyroidectomy is warranted.[1] Patients with early stage I and stage II disease can achieve 100% survival rates regardless of the ATA risk category.[104] Common pathogenic variants in the ATA-MOD category are shown in Table 5.
Pathogenic variants at codons 883 and 918 have been seen only in MEN2B and are associated with the earliest age of onset and the most aggressive form of MTC.[103,105,106,107,108,109] Approximately 95% of individuals with MEN2B will have the M918T pathogenic variant.[105,106,107,110] As discussed above, 50% of individuals with MEN2B will develop PHEO but PHPT is rare. A retrospective review of all published cases of A883F variant carriers (N = 13) found that the MTC disease course was more indolent than what was observed in M918T carriers. A883F carriers had later disease onset (50% penetrance for MTC at age 19 y), 5- and 10-year survival rates of 88%, and 63% of patients achieved biochemical cure for MTC.[103] In addition to variants at codons 883 and 918, some individuals with an MEN2B-like phenotype have been found to carry two germline variants.[111,112,113,114,115] It is likely that as testing for RET becomes more common in clinical practice, additional double variant phenotypes will be described.
Pathogenic variants at codon 634 (ATA-H) are by far the most frequent finding in families with MEN2A. One study of 477 RET carriers showed that 52.1% had the C634R pathogenic variant, 26.0% carried the C634Y pathogenic variant, and 9.1% had the C634G pathogenic variant.[49] In general, pathogenic variants at codon 634 are associated with PHEOs and PHPT.[49,116] Until recently, MEN2A with cutaneous lichen amyloidosis had been seen almost exclusively in patients with pathogenic variants at codon 634.[49,51,117] However, a recent report described MTC and cutaneous lichen amyloidosis in an individual previously thought to have FMTC due to a codon 804 pathogenic variant.[118] Codon 634 pathogenic variants have also been described in FMTC but are almost exclusively C634Y.[49]
In summary, ATA-HST and ATA-H (previously levels D and C, respectively) pathogenic variants confer the highest risk of MTC (about 95% lifetime risk) with a more aggressive disease course. There is an increased risk of PHEO (up to 50%).[49,119] Individuals with codon 634 pathogenic variants (but not codon 883 or 918 variants) also have an increased risk of PHPT.[49]
Moderate-risk variants located in exon 10 of the RET gene include variants at codons 609, 611, 618, 620, and 630. These variants involve cysteine residues in the extracellular domain of the RET protein and have been seen in families with MEN2A and those with MTC only (FMTC).[21,49,100,120,121,122,123,124] The risk of MTC in individuals with these pathogenic variants is approximately 95% to 100%; the risk of PHEO and hyperparathyroidism is lower than that seen in individuals with high-risk pathogenic variants.
Individuals with pathogenic variants previously classified as ATA-level A (now classified with ATA-level B as ATA-MOD, i.e., codons 321, 515, 533, 600, 603, 606, 531/9 base pair duplication, and 532 duplication) have a lower, albeit still elevated, lifetime risk of MTC. MTC associated with these pathogenic variants tends to follow a more indolent course and have a later age at onset, although there are several reports of individuals with these pathogenic variants who developed MTC before age 20 years.[49,125,126,127,128,129] Although PHEO and PHPT are not commonly associated with these pathogenic variants, they have been described.[129]
| RETPathogenic variant | Exon | Risk of Aggressive MTC | Approximate Incidence of PHEO | Approximate Incidence of HPTH | Presence of CLA | Presence of HSCR |
|---|---|---|---|---|---|---|
| CLA = cutaneous lichen amyloidosis; HSCR = Hirschsprung disease; HPTH = hyperparathyroidism; MTC = medullary thyroid cancer; PHEO = pheochromocytoma. | ||||||
| a Adapted from Wells et al.[1] | ||||||
| G533C | 8 | Moderate | 10% | - | N | N |
| C609F/G/R/S/Y | 10 | Moderate | 10%–30% | 10% | N | Y |
| C611F/G/S/Y/W | 10 | Moderate | 10%–30% | 10% | N | Y |
| C618F/R/S | 10 | Moderate | 10%–30% | 10% | N | Y |
| C620F/R/S | 10 | Moderate | 10%–30% | 10% | N | Y |
| C630R/Y | 11 | Moderate | 10%–30% | 10% | N | N |
| D631Y | 11 | Moderate | 50% | - | N | N |
| C634F/G/R/S/W/Y | 11 | High | 50% | 20%–30% | Y | N |
| K666E | 11 | Moderate | 10% | - | N | N |
| E768D | 13 | Moderate | - | - | N | N |
| L790F | 13 | Moderate | 10% | - | N | N |
| V804L | 14 | Moderate | 10% | 10% | N | N |
| V804M | 14 | Moderate | 10% | 10% | Y | N |
| A883F | 15 | High | 50% | - | N | N |
| S891A | 15 | Moderate | 10% | 10% | N | N |
| R912P | 16 | Moderate | - | - | N | N |
| M918T | 16 | Highest | 50% | - | N | N |
In addition to the pathogenic variants categorized in Table 5, a number of rare or novel RET variants have been described. Some of these represent pathogenic variants that lead to MEN2A phenotypes. Others may represent low-penetrance alleles or modifying alleles that confer only a modest risk of developing MTC.[130] A multicenter study identified eight families with a RET K666N variant. Of the 16 screened family members identified as having a pathogenic variant, only one had MTC.[130] Still others may be benign polymorphisms of no clinical significance. For example, some studies show compelling evidence that RET variants Y791F (p.Tyr791Phe) and S649L (p.Ser649Leu) are likely benign polymorphisms; this was based on the equal frequencies seen between cases and healthy controls and co-occurrence with other disease-causing variants that cosegregate with disease in the family.[131,132] A long-term follow-up study of Danish Y791F carriers (n = 20) showed no sign of MEN2A (MTC, PHPT, PHEO, cutaneous lichen amyloidosis, or HSCR) among the cohort, with a median age of 49.5 years (range, 7–87 y).[133] Therefore, carriers of these variants are not followed with MEN2 management guidelines; asymptomatic family members are generally not tested for these variants. Comprehensive testing of all hotspot variants in exon 8 and exons 10–16 may be performed to rule out any other RET pathogenic variants. However, a PHEO diagnosis may warrant extensive testing of other disease-related genes. For more information, see the Familial Pheochromocytoma and Paraganglioma Syndrome section.
Research is ongoing into the role of neutral RET sequence variants in modifying the clinical presentation of patients with MEN2A. The presence of certain RET polymorphisms or haplotypes is being analyzed for its impact on the likelihood for development of PHEO, hyperparathyroidism, HSCR, and age at onset of metastatic involvement with MTC.[134,135,136,137] A variety of approaches, including segregation analyses, in silico analyses, association studies, and functional assays, can be employed to determine the functional and clinical significance of a given genetic variant. A publicly available RET variant online database repository was developed and includes a complete list of variants and their associated pathogenicity, phenotype, and other associated clinical information and literature references.[138]
Surveillance
Screening at-risk individuals for PHEO
The presence of a functioning PHEO can be excluded by appropriate biochemical screening before thyroidectomy in any patient with MEN2A or MEN2B. However, childhood PHEOs are rare in MEN2.[1] The ATA recommends that annual screening for PHEO be considered by age 11 years in patients with ATA-HST or ATA-H RET pathogenic variants.[1] The ATA recommends that patients with ATA-MOD RET pathogenic variants have periodic screening for PHEO beginning by age 16 years.[1] MRI or other imaging tests may be ordered only if the biochemical results are abnormal.[27,139] Studies of individuals with sporadic or hereditary PHEO (including, but not limited to, individuals with MEN2) have suggested that measurement of catecholamine metabolites, specifically plasma-free metanephrines and/or urinary fractionated metanephrines, provides a higher diagnostic sensitivity than urinary catecholamines because of the episodic nature of catecholamine excretion.[33,34,35,36,37,38,39,140] Several reviews provide a succinct summary of the biochemical diagnosis, localization, and management of PHEO.[39,141] In addition to surgery, there are other clinical situations in which patients with catecholamine excess face special risk. An example is the healthy at-risk female patient who becomes pregnant. Pregnancy, labor, or delivery may precipitate a hypertensive crisis in individuals who have an unrecognized PHEO. Pregnant patients who are found to have catecholamine excess require appropriate pharmacotherapy before delivery.
Level of evidence: 5
Screening at-risk individuals for hyperparathyroidism
MEN2-related hyperparathyroidism is generally associated with mild, often asymptomatic hypercalcemia early in the natural history of the disease, which, left untreated, may become symptomatic.[60] Childhood hyperparathyroidism is rare in MEN2. Three studies found the median age at diagnosis was about 38 years.[60,142,143] The ATA provides recommendations for annual screening for hyperparathyroidism,[1] with screening beginning by age 11 years in carriers of ATA-HST and ATA-H pathogenic variants and by age 16 years for carriers of ATA-MOD RET pathogenic variants.[1] Testing typically includes albumin-corrected calcium or ionized serum calcium with or without intact parathyroid hormone (PTH) measurement.
Level of evidence: 5
Screening at-risk individuals in kindreds without an identifiableRETpathogenic variant
Risk-reducing thyroidectomy is not routinely offered to at-risk individuals unless MEN2A is confirmed. The screening protocol for MTC in patients with MEN2A is annual calcitonin stimulation test; however, caution must be used in interpreting test results because CCH that is not a precursor to MTC occurs in about 5% of the population.[13,14,144] In addition, there is significant risk of false-negative test results in patients younger than 15 years.[14] Screening for PHEO and parathyroid disease is the same as described above.
For patients at risk of FMTC, annual screening for MTC is the same as for patients with MEN2A.
Level of evidence: 5
Interventions
Treatment for MTC
Risk-reducing thyroidectomy
Risk-reducing thyroidectomy (also referred to as early thyroidectomy and previously referred to as prophylactic thyroidectomy) is the oncologic treatment of choice for patients with MEN2. Children with the M918T RET pathogenic variant may benefit from a thyroidectomy in the first year of life, perhaps in the first months of life.[1] Likewise, children with ATA-H category RET pathogenic variants may undergo prophylactic thyroidectomy at age 5 years or earlier, on the basis of serum calcitonin levels. The ATA recommends that children in the ATA-MOD category have a physical examination, ultrasonography of the neck, and measurement of the serum calcitonin beginning around age 5 years as these tumors may have later onset but are similarly aggressive once this is taken into account.[1,102] The absence of an abnormal calcitonin level may prompt continued measurement every 6 to 12 months.
A multidisciplinary team caring for the patient, including the pediatrician, pediatric endocrinologist, and surgeon should determine the timing of surgery in conjunction with the child's parents on the basis of the trend in serum calcitonin levels, ultrasonographic findings, preference of the family, and experience of the treating physicians.[1]
In children with some ATA-H or ATA-MOD RET pathogenic variants, some studies have suggested that basal and pentagastrin-stimulated calcitonin levels could be used to determine the timing of total thyroidectomy.[145,146,147,148] These findings suggest that surgery may be safely delayed in carriers of an RET pathogenic variant until basal or stimulated calcitonin levels increase on routine testing. The benefits of this approach are particularly noteworthy in the younger population of pathogenic variant carriers, as a delay in surgery until the patient is older may reduce the risk of surgical complications. A large study of 2,740 children aged 16 years and under has provided data on age-specific reference ranges for calcitonin levels in younger children that may assist in decision making.[149] Because some calcitonin assays may be more sensitive than others,[147] attention to the type of testing, as well as calcitonin levels will need to be considered. However, normal preoperative basal calcitonin does not exclude the possibility of the patient having MTC.[150]
For patients with RET germline variants, older age at prophylactic thyroidectomy has been significantly associated with a higher risk of postoperative persistent or recurrent disease.[151] Consistent with this, a study of young, clinically asymptomatic individuals with MEN2A showed there was a lower incidence of persistent or recurrent disease in patients who had thyroidectomy earlier in life (defined as younger than age 8 y) and who had no evidence of lymph node metastases.[152] Several studies have shown that there is a significantly lower rate of invasive or metastatic MTC among those who undergo surgery at an earlier age than among those who undergo surgery at a later age.[153] For patients with the most aggressive M918T RET variant, cure is exceptional if surgery is performed after age 4 years.[145,154] Together, these findings are consistent with more favorable outcomes for patients undergoing risk-reducing thyroidectomy at a young age.[155]
Although thyroidectomy before biochemical evidence of disease (normal preoperative calcitonin) may reduce the risk of recurrent disease, a selective strategy for postoperative and lifelong surveillance might depend on the final pathologic determination of whether a carcinoma was present and whether it was micromedullary or macromedullary.[1,156] One study found that 10% of patients with MEN2A undergoing thyroidectomy developed recurrent disease, on the basis of initially undetectable basal and stimulated calcitonin levels (<2 pg/mL) that became positive 5 to 10 years after surgery.[152] Only 2% of patients had residual disease after prophylactic surgery as assessed by a persistently elevated basal or stimulated calcitonin.[152]
Questions remain concerning the natural history of MEN2. As more information is acquired, recommendations regarding the optimal age for thyroidectomy and the potential role for genetics and biochemical screening may change. Earliest reports of MTC in MEN2B before age 3 years, and before age 6 years in MEN2A cases with ATA-H or ATA-MOD RET variants have been documented.[145,152,154,157] Conversely, another case report documented onset of cancer in midlife or later in some families with the FMTC subtype of MEN2A and in elderly relatives who carry the RET variant genotype but have not developed cancer.[158] Subsequent data have suggested that some ATA-MOD RET variants, which were previously thought of as more indolent may be as aggressive as ATA-H variants, but are associated with delayed onset of disease.[102,104] Emerging data pertaining to the timing of disease onset and local metastases by risk category are evolving.[104] These clinical observations suggest that the natural history of the MEN2 syndromes is variable and could be subject to modifying effects related to specific RET pathogenic variants, other genes, behavioral factors, or environmental exposures.
Level of evidence: 4b
Therapeutic thyroidectomy
The standard treatment for adults with MTC is surgical removal of the entire thyroid gland, including the posterior capsule and central lymph node dissection.[1] A therapeutic central neck dissection is typically performed if there is radiographic evidence of metastatic lymph node involvement or if the serum calcitonin level is higher than 40 pg/mL.[1] The decision to perform a prophylactic central neck dissection is generally made on the basis of multiple factors such as patient age, pathogenic variant, presence of concomitant PHPT, and the vascularity of the parathyroid glands.[1] Selective autotransplantation of parathyroid glands that were devascularized during a prophylactic thyroidectomy and/or central neck clearance is recommended. A selective approach also significantly reduces the detrimental outcome of hypoparathyroidism.[159]
The MEN2B RET variant M918T is associated with approximately 100% incidence of MTC in the first years of life [154] and is considered the most aggressive MEN2 phenotype. In patients with MEN2A, the ATA-H high-risk codon 634 pathogenic variant is much more likely to be associated with invasive or metastatic MTC and development of persistent or recurrent disease than pathogenic variants in codons 804, 618, or 620.[153] One series of 503 at-risk individuals with ATA-MOD category pathogenic variants (including codons 533, 609, 611, 618, 620, 791, and 804) reported cumulative penetrance rates, median time to MTC, and positive predictive value of preoperative calcitonin.[146] The risk of developing MTC by age 50 years ranged from 18% to 95%, depending on the codon, with codon 620 having the highest penetrance. Most patients with MTC had node-negative disease, confirming the more indolent disease course that had been previously reported with these pathogenic variants. Although an elevated preoperative calcitonin level strongly predicted the presence of MTC, relatively high false-negative rates (low normal calcitonin levels with MTC) were noted for many of the codons. This information is useful when counseling carriers of pathogenic variants regarding the extent of surgical resection.
The ATA recommends compartment-directed lymph node dissection for local or regional disease (no evidence of distant metastases) in the following situations:[1]
- If there is no evidence of neck nodal metastases by ultrasonography in biopsy-proven thyroid disease, prophylactic central neck dissection should be performed concomitant with initial thyroidectomy.
- If nodal disease is present in either the central or lateral neck, a compartment-oriented lateral neck dissection of the ipsilateral side should be performed.
- If nodal disease is present and basal calcitonin levels are greater than 200 pg/mL, then consider contralateral lateral neck dissection.
Although basal calcitonin levels may not be able to identify all patients with MTC preoperatively, this test has utility as a predictor of postoperative remission, lymph node metastases, and distant metastases.[160] In one study of 224 patients from a single institution, preoperative basal calcitonin levels greater than 500 pg/mL predicted failure to achieve biochemical remission.[160] The authors of this study found that nodal metastases started appearing at basal calcitonin levels of 40 pg/mL (normal, <10 pg/mL). In node-positive patients, distant metastases emerged at basal calcitonin levels of 150 pg/mL to 400 pg/mL. Another study of 308 RET pathogenic variant carriers found that a normal basal preoperative calcitonin excluded the presence of lymph node metastases (negative predictive value, 100%).[148] Therefore, the preoperative basal calcitonin level is a useful prognostic indicator and may help guide the surgical approach.
With regard to prognosis, structural and metastatic disease recurrence is common in germline RET pathogenic variant carriers and can happen up to 20 years after initial treatment. Despite this, overall survival (OS) is generally favorable, with one study citing an OS rate of 92% at 10 years.[161]
Patients who have had total thyroidectomy will require lifelong thyroid hormone replacement therapy. The dosing of medication is age-dependent, and treatment may be initiated on the basis of ideal body weight. For healthy adults aged 60 years and younger with no cardiac disease, a reasonable starting dose is 1.6 µg/kg to 1.8 µg/kg given once daily.[162] Older patients may require 20% to 30% less thyroid hormone.[163] Children metabolize T4 more rapidly than adults and consequently require relatively higher replacement by body weight. Depending on the age of the child, replacement is typically between 2 µg/kg to 6 µg/kg.[164] It is important to note, however, that replacement is preferred over suppressive therapy. Since C-cell tumors are not thyroid-stimulating hormone (TSH)–dependent for growth, the T4 therapy for patients with MTC therefore may be adjusted to maintain a TSH within the normal reference range. Thyroglobulin measurement may also be useful for adjusting and maintaining TSH levels within a normal reference range to prevent additional regrowth of remnant thyroid tissue.[165]
Level of evidence (central neck dissection): 5
Level of evidence (hormone replacement therapy): 3c
Level of evidence (therapeutic thyroidectomy): 4
Adjuvant therapy
Chemotherapy and radiation therapy are generally not effective against MTC.[4,166,167] Hence, targeted molecular therapies are being explored to manage MTC. RET inhibitors and multikinase inhibitors are being used to block RET activity.
There are two U.S. Food and Drug Administration–approved RET inhibitors (pralsetinib and selpercatinib) that are now available for patients with MTC who have a RETpoint mutation. These RET inhibitors are also available for patients who have differentiated thyroid cancers with a RET fusion. A multicenter, phase I/II trial (ARROW) was conducted to evaluate the efficacy of pralsetinib in patients with RET-mutant MTC with or without prior treatment with vandetanib or cabozantinib. Among 55 patients who were previously treated with a multikinase inhibitor, the overall response rate was 60% (95% confidence interval [CI], 46%–73%) and 1-year progression-free survival (PFS) rate was 75% (95% CI, 63%–86%). Among 21 treatment-naïve patients, the overall response rate was 71% (95% CI, 48%–89%) and the 1-year PFS rate was 81% (95% CI, 63%–98%).[168] A similar phase I/II trial (LIBRETTO) examined the efficacy of selpercatinib in patients with RET-mutant MTC with or without prior treatment with vandetanib or cabozantinib. Among 55 patients who were previously treated with a multikinase inhibitor, 69% had an objective response (95% CI, 55%–81%); the 1-year PFS rate was 82% (95% CI, 69%–90%). Among 88 treatment-naïve patients, the objective response rate was 73% (95% CI, 62%–82%) and the 1-year PFS rate was 92% (95% CI, 82%–97%).[169]
The use of vandetanib and cabozantinib is approved by the U.S. Food and Drug Administration for adult patients with progressive metastatic MTC who are ineligible for surgery. A phase III study found that progression-free survival (PFS) was longer in adults who received vandetanib than in those who received placebo.[170] A phase I/phase II study of children with MEN2B found an objective partial response rate of 47% with vandetanib.[171] Subsequent follow-up analysis of this cohort revealed that a partial response was seen in 10 of 17 patients; stable disease was seen in an additional 6 individuals. Median PFS was 6.7 years.[172] A double-blind phase III trial that compared cabozantinib with placebo in 330 patients with progressive MTC showed an improvement in median PFS across all subgroups.[173,174] In this trial, patients who had pathogenic variants, including RET or RAS, were more likely to have a prolonged PFS compared with patients lacking both pathogenic variants.[175] Prospective studies may further clarify whether particular pathogenic variants can be used to guide therapy. Neither cabozantinib nor vandetanib has demonstrated improved OS.[170,173,174] Importantly, these agents are not effective at inhibiting some MEN2 RET variants, specifically those at codon 804,[176] making genotype an important consideration for treatment with RET inhibitors. Further, a 2018 study has demonstrated the development of resistance to these agents through somatic acquisition of a V804M mutation in RET.[177] Finally, multikinase inhibitors are associated with significant toxicities, possibly due to their off-target effects on other kinases.[178] Other multikinase inhibitors for targeting RET are being studied in clinical trials; however, they may only provide limited advantages over vandetanib and cabozantinib. For these reasons, ongoing studies are focusing on the development of selective RET inhibitors with fewer off-target effects that are able to block the activity of all RET variant forms and the use of combination therapy in MTC. Future studies will likely focus on the development of new targeted therapies and the use of combination therapy in MTC.[179,180]
Level of evidence (pralsetinib): 4
Level of evidence (selpercatinib): 3dii
Level of evidence (vandetanib): 2
Level of evidence (cabozantinib): 1
For more information, see Thyroid Cancer Treatment.
Treatment for MEN2-related PHEO
A cognitive shift has occurred in the field regarding the risks and benefits of whole organ resection. This is especially relevant for endocrine glands that are difficult to manage postresection and may require replacement therapy. PHEO may be either unilateral or bilateral in patients with MEN2. Laparoscopic adrenalectomy (anterior or posterior) is the recommended approach after appropriate preoperative medical blockade for the treatment of unilateral PHEO.[1,93,101,181] The risks, benefits, and potential of life-threatening adrenal insufficiency should be considered at the time of the initial operative planning. If disease appears unilateral, the contralateral gland may develop metachronous disease in 17% to 72% of patients.[6,182] In a series of RET p.Cys634 variant carriers, only 1 in 5 developed a second ipsilateral PHEO after a follow-up period of 8 to 11 years when the cortical preservation technique was used. This technique allows patients to continue making steroid hormones on their own, which makes it a viable surgical option.[183] In one series, 23 patients with a unilateral PHEO and a macroscopically normal contralateral adrenal gland were treated initially with unilateral adrenalectomy.[184] A PHEO developed within the retained gland in 12 (52%) of these patients, occurring a mean of 11.9 years after initial surgery. During follow-up, no patient experienced a hypertensive crisis or other problems attributable to an undiagnosed PHEO. In contrast, 10 of 43 patients (23%) treated with bilateral adrenalectomy experienced at least one episode of acute adrenal insufficiency. Thus, unilateral adrenalectomy is a reasonable management strategy for unilateral PHEO in patients with MEN2.[1,185,186] Many experts suggest considering a cortical-sparing technique for a suspected unilateral PHEO, even if it is the patient's first operation.[1,187] For more information, see the section on Interventions in the Familial Pheochromocytoma and Paraganglioma Syndrome section. Because of the risk of contralateral gland disease, periodic surveillance (serum or urinary catecholamine measurements) for the development of disease in the contralateral adrenal gland is recommended.[1]
Regarding the operative approach, several studies examined the value of a posterior retroperitoneoscopic adrenalectomy and found it to be safe and effective, with very low mortality and a low rate of minor complications, and conversion to open surgery required rarely.[182,188,189,190,191,192,193,194]
Level of evidence: 4
Treatment for hyperparathyroidism
Most patients with MEN2-related parathyroid disease are either asymptomatic or diagnosed incidentally in the preoperative planning or at the time of thyroidectomy. Typically, the hypercalcemia (when present) is mild, although it may be associated with increased urinary excretion of calcium and nephrolithiasis. As a consequence, the indications for surgical intervention are generally similar to those recommended for patients with sporadic PHPT.[93] In general, fewer than four of the parathyroid glands are involved at the time of detected abnormalities in calcium metabolism.[1]
Treatment of hyperparathyroidism typically employs some extent of surgical removal of the involved glands. Cure of hyperparathyroidism was achieved surgically in 89% of one large series of patients;[60] however, 22% of resected patients in this study developed postoperative hypoparathyroidism. Five patients (9%) had recurrent hyperparathyroidism. This series employed various surgical techniques, including total parathyroidectomy with autotransplantation to the nondominant forearm (4 of 11 patients [36%] developed postoperative hypoparathyroidism), subtotal parathyroidectomy (6 of 12 patients [50%] developed hypoparathyroidism), and resection only of glands that were macroscopically enlarged (3 of 29 patients [10%] developed hypoparathyroidism). These data indicate that excision of only those parathyroid glands that are enlarged appears to be sufficient therapy in most cases.
Some investigators have suggested using the MEN2 subtype to decide where to place the parathyroid glands that are identified at the time of thyroid surgery. For patients with MEN2B in whom the risk of parathyroid disease is quite low, the parathyroid glands may be left in situ in the neck. For adult patients with MEN2A, in whom the glands have been inadvertently devascularized during primary surgical treatment for MTC, it is suggested that the glands needing reimplantation be implanted in the nondominant forearm. This approach minimizes the need for further surgical intervention in the neck should hyperparathyroidism develop or recur.[1,159,195,196] For children, the risk/benefit ratio must be carefully weighed to avoid overtreatment and subsequent aparathyroidism.[197] It is important to confirm that the remnant or autotransplanted parathyroid tissue is functional.[1,101,198,199]
Medical therapy of hyperparathyroidism has gained popularity with the advent of calcimimetics, agents that sensitize the calcium-sensing receptors on the parathyroid glands to circulating calcium levels and thereby reduce circulating PTH levels. In a randomized, double-blind, placebo-controlled trial, cinacalcet hydrochloride was shown to induce sustained reduction in circulating calcium and PTH levels in patients with PHPT.[200] In patients who are high-risk surgical candidates, those with recurrent hyperparathyroidism, or those in whom life expectancy is limited, medical therapy may be a viable alternative to a surgical approach.[1] Consequences of long-term therapy with cinacalcet are unknown.
Level of evidence: 5
Genetic Counseling
Mode of inheritance
All of the MEN2 subtypes are inherited in an autosomal dominant manner. For the child of someone with MEN2, the risk of inheriting the MEN2 pathogenic variant is 50%. Some individuals with MEN2, however, carry a de novo pathogenic variant; that is, they carry a new pathogenic variant that was not present in previous generations of their family and thus do not have an affected parent. The proportion of individuals with MEN2 who have an affected parent varies by subtype.
MEN2A: About 95% of affected individuals have an affected parent. It is appropriate to evaluate the parents of an individual with MEN2A for manifestations of the disorder. In the 5% of cases that are not familial, either de novo pathogenic variants or incomplete penetrance of the mutant allele is possible.[201]
FMTC: Multiple family members are affected; therefore, all affected individuals inherited the mutant gene from a parent.
MEN2B: About 50% of affected individuals have de novo RET gene pathogenic variants, and 50% have inherited the pathogenic variant from a parent.[202,203] The majority of de novo pathogenic variants are paternal in origin, but cases of maternal origin have been reported.[204]
Siblings of a proband: The risk to siblings depends on the genetic status of the parent, which can be clarified by pedigree analysis and/or DNA-based testing. In situations of apparent de novo pathogenic variants, germline mosaicism in an apparently unaffected parent must be considered, even though such an occurrence has not yet been reported.
Attitudes toward preimplantation genetic testing
One study explored the attitudes of individuals with MEN1 and MEN2 toward preimplantation genetic testing (PGT).[205] Ninety-one clinic-based patients from a single U.S. institution who had MEN1 and an MEN1 pathogenic variant or MEN2 and a RET pathogenic variant were surveyed. The study found that 30% (10 of 33) of those with MEN1 and 37% (21 of 57) of those with MEN2 were aware of PGT; 82% (27 of 33) of those with MEN1 and 61% (34 of 56) of those with MEN2 thought PGT should be offered; and 61% (19 of 31) of those with MEN1 and 43% (23 of 54) of those with MEN2 would consider PGT.
Psychosocial issues
The psychosocial impact of genetic testing for pathogenic variants in RET has not been extensively studied. Published studies have had limitations such as small sample size and heterogeneous populations; thus, the clinical relevance of these findings should be interpreted with caution. Identification as the carrier of a pathogenic variant may affect self-esteem, family relationships, and quality of life.[206] In addition, misconceptions about genetic disease may result in familial blame and guilt.[207,208] Several review articles outline both the medical and psychological issues, especially those related to the testing of children.[209,210,211,212] The medical value of early screening and risk-reducing treatment are contrasted with the loss of decision-making autonomy for the individual. Lack of agreement between parents about the value and timing of genetic testing and surgery may spur the development of emotional problems within the family.
One study examined levels of psychological distress in the interval between submitting a blood sample and receiving genetic test results. Those individuals who experienced the highest level of distress were younger than 25 years, single, and had a history of responding to distressful situations with anxiety.[213] Pathogenic variant–positive parents whose children received negative test results did not seem to be reassured, questioned the reliability of the DNA test, and were eager to continue screening of their noncarrier children.[214]
A small qualitative study (N = 21) evaluated how patients with MEN2A and family members conceptualized participation in lifelong high-risk surveillance.[215] Ongoing surveillance was viewed as a reminder of a health threat. Acceptance and incorporation of lifelong surveillance into routine health care was essential for coping with the implications of this condition. Concern about genetic predisposition to cancer was peripheral to concerns about surveillance. Supportive interventions, such as Internet discussion forums, can serve as an ongoing means of addressing informational and support needs of patients with MTC undergoing lifelong surveillance.[216]
References:
- Wells SA, Asa SL, Dralle H, et al.: Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25 (6): 567-610, 2015.
- Kaserer K, Scheuba C, Neuhold N, et al.: Sporadic versus familial medullary thyroid microcarcinoma: a histopathologic study of 50 consecutive patients. Am J Surg Pathol 25 (10): 1245-51, 2001.
- Robbins J, Merino MJ, Boice JD, et al.: Thyroid cancer: a lethal endocrine neoplasm. Ann Intern Med 115 (2): 133-47, 1991.
- Moley JF, Debenedetti MK, Dilley WG, et al.: Surgical management of patients with persistent or recurrent medullary thyroid cancer. J Intern Med 243 (6): 521-6, 1998.
- Machens A, Lorenz K, Weber F, et al.: Exceptionality of Distant Metastasis in Node-Negative Hereditary and Sporadic Medullary Thyroid Cancer: Lessons Learned. J Clin Endocrinol Metab 106 (8): e2968-e2979, 2021.
- Thosani S, Ayala-Ramirez M, Palmer L, et al.: The characterization of pheochromocytoma and its impact on overall survival in multiple endocrine neoplasia type 2. J Clin Endocrinol Metab 98 (11): E1813-9, 2013.
- Machens A, Lorenz K, Dralle H: Constitutive RET tyrosine kinase activation in hereditary medullary thyroid cancer: clinical opportunities. J Intern Med 266 (1): 114-25, 2009.
- DeLellis RA, Lloyd RV, Heitz PU, et al., eds.: Pathology and Genetics of Tumours of Endocrine Organs. IARC Press, 2004. World Health Organization classification of tumours, vol. 8.
- Guyétant S, Rousselet MC, Durigon M, et al.: Sex-related C cell hyperplasia in the normal human thyroid: a quantitative autopsy study. J Clin Endocrinol Metab 82 (1): 42-7, 1997.
- LiVolsi VA: C cell hyperplasia/neoplasia. J Clin Endocrinol Metab 82 (1): 39-41, 1997.
- Mete O, Asa SL: Precursor lesions of endocrine system neoplasms. Pathology 45 (3): 316-30, 2013.
- Elisei R, Bottici V, Luchetti F, et al.: Impact of routine measurement of serum calcitonin on the diagnosis and outcome of medullary thyroid cancer: experience in 10,864 patients with nodular thyroid disorders. J Clin Endocrinol Metab 89 (1): 163-8, 2004.
- Landsvater RM, Rombouts AG, te Meerman GJ, et al.: The clinical implications of a positive calcitonin test for C-cell hyperplasia in genetically unaffected members of an MEN2A kindred. Am J Hum Genet 52 (2): 335-42, 1993.
- Lips CJ, Landsvater RM, Höppener JW, et al.: Clinical screening as compared with DNA analysis in families with multiple endocrine neoplasia type 2A. N Engl J Med 331 (13): 828-35, 1994.
- Kudo T, Miyauchi A, Ito Y, et al.: Serum calcitonin levels with calcium loading tests before and after total thyroidectomy in patients with thyroid diseases other than medullary thyroid carcinoma. Endocr J 58 (3): 217-21, 2011.
- Howlader N, Noone AM, Krapcho M, et al.: SEER Cancer Statistics Review (CSR) 1975-2017. Bethesda, Md: National Cancer Institute, 2020. Available online. Last accessed February 10, 2023.
- Gharib H, McConahey WM, Tiegs RD, et al.: Medullary thyroid carcinoma: clinicopathologic features and long-term follow-up of 65 patients treated during 1946 through 1970. Mayo Clin Proc 67 (10): 934-40, 1992.
- Ponder BA: Multiple endocrine neoplasia type 2. In: Vogelstein B, Kinzler KW, eds.: The Genetic Basis of Human Cancer. 2nd ed. McGraw-Hill, 2002, pp 501-513.
- Decker RA, Peacock ML, Borst MJ, et al.: Progress in genetic screening of multiple endocrine neoplasia type 2A: is calcitonin testing obsolete? Surgery 118 (2): 257-63; discussion 263-4, 1995.
- Kitamura Y, Goodfellow PJ, Shimizu K, et al.: Novel germline RET proto-oncogene mutations associated with medullary thyroid carcinoma (MTC): mutation analysis in Japanese patients with MTC. Oncogene 14 (25): 3103-6, 1997.
- Eng C, Mulligan LM, Smith DP, et al.: Low frequency of germline mutations in the RET proto-oncogene in patients with apparently sporadic medullary thyroid carcinoma. Clin Endocrinol (Oxf) 43 (1): 123-7, 1995.
- Wohllk N, Cote GJ, Bugalho MM, et al.: Relevance of RET proto-oncogene mutations in sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab 81 (10): 3740-5, 1996.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Thyroid Carcinoma. Version 2.2023. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free subscription Last accessed May 25, 2023.
- American Cancer Society: Cancer Facts and Figures 2023. American Cancer Society, 2023. Available online. Last accessed June 8, 2023.
- Randle RW, Balentine CJ, Leverson GE, et al.: Trends in the presentation, treatment, and survival of patients with medullary thyroid cancer over the past 30 years. Surgery 161 (1): 137-146, 2017.
- Hundahl SA, Fleming ID, Fremgen AM, et al.: A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985-1995 [see comments] Cancer 83 (12): 2638-48, 1998.
- Modigliani E, Vasen HM, Raue K, et al.: Pheochromocytoma in multiple endocrine neoplasia type 2: European study. The Euromen Study Group. J Intern Med 238 (4): 363-7, 1995.
- Bhattacharyya N: A population-based analysis of survival factors in differentiated and medullary thyroid carcinoma. Otolaryngol Head Neck Surg 128 (1): 115-23, 2003.
- Roman S, Lin R, Sosa JA: Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer 107 (9): 2134-42, 2006.
- Bergholm U, Bergström R, Ekbom A: Long-term follow-up of patients with medullary carcinoma of the thyroid. Cancer 79 (1): 132-8, 1997.
- Kebebew E, Ituarte PH, Siperstein AE, et al.: Medullary thyroid carcinoma: clinical characteristics, treatment, prognostic factors, and a comparison of staging systems. Cancer 88 (5): 1139-48, 2000.
- Romei C, Ciampi R, Elisei R: A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol 12 (4): 192-202, 2016.
- Lenders JW, Pacak K, Walther MM, et al.: Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 287 (11): 1427-34, 2002.
- Gerlo EA, Sevens C: Urinary and plasma catecholamines and urinary catecholamine metabolites in pheochromocytoma: diagnostic value in 19 cases. Clin Chem 40 (2): 250-6, 1994.
- Guller U, Turek J, Eubanks S, et al.: Detecting pheochromocytoma: defining the most sensitive test. Ann Surg 243 (1): 102-7, 2006.
- Raber W, Raffesberg W, Bischof M, et al.: Diagnostic efficacy of unconjugated plasma metanephrines for the detection of pheochromocytoma. Arch Intern Med 160 (19): 2957-63, 2000.
- Sawka AM, Jaeschke R, Singh RJ, et al.: A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 88 (2): 553-8, 2003.
- Unger N, Pitt C, Schmidt IL, et al.: Diagnostic value of various biochemical parameters for the diagnosis of pheochromocytoma in patients with adrenal mass. Eur J Endocrinol 154 (3): 409-17, 2006.
- Pacak K, Eisenhofer G, Ahlman H, et al.: Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab 3 (2): 92-102, 2007.
- van der Harst E, de Herder WW, Bruining HA, et al.: [(123)I]metaiodobenzylguanidine and [(111)In]octreotide uptake in begnign and malignant pheochromocytomas. J Clin Endocrinol Metab 86 (2): 685-93, 2001.
- Pacak K, Linehan WM, Eisenhofer G, et al.: Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med 134 (4): 315-29, 2001.
- Kaelin WG: Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer 2 (9): 673-82, 2002.
- Maher ER, Eng C: The pressure rises: update on the genetics of phaeochromocytoma. Hum Mol Genet 11 (20): 2347-54, 2002.
- Fraser WD: Hyperparathyroidism. Lancet 374 (9684): 145-58, 2009.
- Tonelli F, Marcucci T, Giudici F, et al.: Surgical approach in hereditary hyperparathyroidism. Endocr J 56 (7): 827-41, 2009.
- Villablanca A, Calender A, Forsberg L, et al.: Germline and de novo mutations in the HRPT2 tumour suppressor gene in familial isolated hyperparathyroidism (FIHP). J Med Genet 41 (3): e32, 2004.
- Marx SJ, Simonds WF, Agarwal SK, et al.: Hyperparathyroidism in hereditary syndromes: special expressions and special managements. J Bone Miner Res 17 (Suppl 2): N37-43, 2002.
- Sipple JH: The association of pheochromocytoma with carcinoma of the thyroid gland. Am J Med 31 (1): 163-166, 1961.
- Eng C, Clayton D, Schuffenecker I, et al.: The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 276 (19): 1575-9, 1996.
- Sanso GE, Domene HM, Garcia R, et al.: Very early detection of RET proto-oncogene mutation is crucial for preventive thyroidectomy in multiple endocrine neoplasia type 2 children: presence of C-cell malignant disease in asymptomatic carriers. Cancer 94 (2): 323-30, 2002.
- Yip L, Cote GJ, Shapiro SE, et al.: Multiple endocrine neoplasia type 2: evaluation of the genotype-phenotype relationship. Arch Surg 138 (4): 409-16; discussion 416, 2003.
- Rambaud JC, Jian R, Flourié B, et al.: Pathophysiological study of diarrhoea in a patient with medullary thyroid carcinoma. Evidence against a secretory mechanism and for the role of shortened colonic transit time. Gut 29 (4): 537-43, 1988.
- Cox TM, Fagan EA, Hillyard CJ, et al.: Rôle of calcitonin in diarrhoea associated with medullary carcinoma of the thyroid. Gut 20 (7): 629-33, 1979.
- Raue F, Frank-Raue K, Grauer A: Multiple endocrine neoplasia type 2. Clinical features and screening. Endocrinol Metab Clin North Am 23 (1): 137-56, 1994.
- Perren A, Komminoth P: Familial pheochromocytomas and paragangliomas: stories from the sign-out room. Endocr Pathol 17 (4): 337-44, 2006.
- Webb TA, Sheps SG, Carney JA: Differences between sporadic pheochromocytoma and pheochromocytoma in multiple endocrime neoplasia, type 2. Am J Surg Pathol 4 (2): 121-6, 1980.
- Lips KJ, Van der Sluys Veer J, Struyvenberg A, et al.: Bilateral occurrence of pheochromocytoma in patients with the multiple endocrine neoplasia syndrome type 2A (Sipple's syndrome). Am J Med 70 (5): 1051-60, 1981.
- Neumann HP, Berger DP, Sigmund G, et al.: Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med 329 (21): 1531-8, 1993.
- Conte-Devolx B, Schuffenecker I, Niccoli P, et al.: Multiple endocrine neoplasia type 2: management of patients and subjects at risk. French Study Group on Calcitonin-Secreting Tumors (GETC). Horm Res 47 (4-6): 221-6, 1997.
- Kraimps JL, Denizot A, Carnaille B, et al.: Primary hyperparathyroidism in multiple endocrine neoplasia type IIa: retrospective French multicentric study. Groupe d'Etude des Tumeurs á Calcitonine (GETC, French Calcitonin Tumors Study Group), French Association of Endocrine Surgeons. World J Surg 20 (7): 808-12; discussion 812-3, 1996.
- Benson L, Ljunghall S, Akerström G, et al.: Hyperparathyroidism presenting as the first lesion in multiple endocrine neoplasia type 1. Am J Med 82 (4): 731-7, 1987.
- Trump D, Farren B, Wooding C, et al.: Clinical studies of multiple endocrine neoplasia type 1 (MEN1) QJM 89 (9): 653-69, 1996.
- Vasen HF, Lamers CB, Lips CJ: Screening for the multiple endocrine neoplasia syndrome type I. A study of 11 kindreds in The Netherlands. Arch Intern Med 149 (12): 2717-22, 1989.
- Bugalho MJ, Limbert E, Sobrinho LG, et al.: A kindred with multiple endocrine neoplasia type 2A associated with pruritic skin lesions. Cancer 70 (11): 2664-7, 1992.
- Robinson MF, Furst EJ, Nunziata V, et al.: Characterization of the clinical features of five families with hereditary primary cutaneous lichen amyloidosis and multiple endocrine neoplasia type 2. Henry Ford Hosp Med J 40 (3-4): 249-52, 1992.
- Romeo G, Ceccherini I, Celli J, et al.: Association of multiple endocrine neoplasia type 2 and Hirschsprung disease. J Intern Med 243 (6): 515-20, 1998.
- Mulligan LM, Eng C, Attié T, et al.: Diverse phenotypes associated with exon 10 mutations of the RET proto-oncogene. Hum Mol Genet 3 (12): 2163-7, 1994.
- Decker RA, Peacock ML, Watson P: Hirschsprung disease in MEN 2A: increased spectrum of RET exon 10 genotypes and strong genotype-phenotype correlation. Hum Mol Genet 7 (1): 129-34, 1998.
- Carrasquillo MM, McCallion AS, Puffenberger EG, et al.: Genome-wide association study and mouse model identify interaction between RET and EDNRB pathways in Hirschsprung disease. Nat Genet 32 (2): 237-44, 2002.
- Fewtrell MS, Tam PK, Thomson AH, et al.: Hirschsprung's disease associated with a deletion of chromosome 10 (q11.2q21.2): a further link with the neurocristopathies? J Med Genet 31 (4): 325-7, 1994.
- Pacini F, Castagna MG, Cipri C, et al.: Medullary thyroid carcinoma. Clin Oncol (R Coll Radiol) 22 (6): 475-85, 2010.
- Brauckhoff M, Machens A, Hess S, et al.: Premonitory symptoms preceding metastatic medullary thyroid cancer in MEN 2B: An exploratory analysis. Surgery 144 (6): 1044-50; discussion 1050-3, 2008.
- Castinetti F, Moley J, Mulligan L, et al.: A comprehensive review on MEN2B. Endocr Relat Cancer 25 (2): T29-T39, 2018.
- Gorlin RJ, Sedano HO, Vickers RA, et al.: Multiple mucosal neuromas, pheochromocytoma and medullary carcinoma of the thyroid--a syndrome. Cancer 22 (2): 293-9 passim, 1968.
- Gorlin RJ, Vickers RA: Multiple mucosal neuromas, pheochromocytoma, medullary carcinoma of the thyroid and marfanoid body build with muscle wasting: reexamination of a syndrome of neural crest malmigration. Birth Defects Orig Artic Ser 7 (6): 69-72, 1971.
- Makri A, Akshintala S, Derse-Anthony C, et al.: Multiple Endocrine Neoplasia Type 2B Presents Early in Childhood but Often Is Undiagnosed for Years. J Pediatr 203: 447-449, 2018.
- Skinner MA, DeBenedetti MK, Moley JF, et al.: Medullary thyroid carcinoma in children with multiple endocrine neoplasia types 2A and 2B. J Pediatr Surg 31 (1): 177-81; discussion 181-2, 1996.
- Wells SA, Pacini F, Robinson BG, et al.: Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: an update. J Clin Endocrinol Metab 98 (8): 3149-64, 2013.
- van den Broek MFM, Rijks EBG, Nikkels PGJ, et al.: Timely diagnosis of multiple endocrine neoplasia 2B by identification of intestinal ganglioneuromatosis: a case series. Endocrine 72 (3): 905-914, 2021.
- Gfroerer S, Theilen TM, Fiegel H, et al.: Identification of intestinal ganglioneuromatosis leads to early diagnosis of MEN2B: role of rectal biopsy. J Pediatr Surg 52 (7): 1161-1165, 2017.
- Hampel H, Bennett RL, Buchanan A, et al.: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 17 (1): 70-87, 2015.
- Bashford MT, Kohlman W, Everett J, et al.: Addendum: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 21 (12): 2844, 2019.
- Gardner E, Papi L, Easton DF, et al.: Genetic linkage studies map the multiple endocrine neoplasia type 2 loci to a small interval on chromosome 10q11.2. Hum Mol Genet 2 (3): 241-6, 1993.
- Mole SE, Mulligan LM, Healey CS, et al.: Localisation of the gene for multiple endocrine neoplasia type 2A to a 480 kb region in chromosome band 10q11.2. Hum Mol Genet 2 (3): 247-52, 1993.
- Takahashi M, Ritz J, Cooper GM: Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 42 (2): 581-8, 1985.
- Kwok JB, Gardner E, Warner JP, et al.: Structural analysis of the human ret proto-oncogene using exon trapping. Oncogene 8 (9): 2575-82, 1993.
- Myers SM, Eng C, Ponder BA, et al.: Characterization of RET proto-oncogene 3' splicing variants and polyadenylation sites: a novel C-terminus for RET. Oncogene 11 (10): 2039-45, 1995.
- Airaksinen MS, Saarma M: The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 3 (5): 383-94, 2002.
- Mulligan LM: RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer 14 (3): 173-86, 2014.
- Robson ME, Bradbury AR, Arun B, et al.: American Society of Clinical Oncology Policy Statement Update: Genetic and Genomic Testing for Cancer Susceptibility. J Clin Oncol 33 (31): 3660-7, 2015.
- Robson ME, Storm CD, Weitzel J, et al.: American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol 28 (5): 893-901, 2010.
- Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. American Society of Human Genetics Board of Directors, American College of Medical Genetics Board of Directors. Am J Hum Genet 57 (5): 1233-41, 1995.
- Brandi ML, Gagel RF, Angeli A, et al.: Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86 (12): 5658-71, 2001.
- Cooper DS, Doherty GM, Haugen BR, et al.: Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 19 (11): 1167-214, 2009.
- Wells SA: Advances in the management of MEN2: from improved surgical and medical treatment to novel kinase inhibitors. Endocr Relat Cancer 25 (2): T1-T13, 2018.
- Romei C, Cosci B, Renzini G, et al.: RET genetic screening of sporadic medullary thyroid cancer (MTC) allows the preclinical diagnosis of unsuspected gene carriers and the identification of a relevant percentage of hidden familial MTC (FMTC). Clin Endocrinol (Oxf) 74 (2): 241-7, 2011.
- Elisei R, Cosci B, Romei C, et al.: Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab 93 (3): 682-7, 2008.
- Sarika HL, Papathoma A, Garofalaki M, et al.: Genetic screening of patients with medullary thyroid cancer in a referral center in Greece during the past two decades. Eur J Endocrinol 172 (4): 501-9, 2015.
- Mathiesen JS, Kroustrup JP, Vestergaard P, et al.: Distribution of RET Mutations in Multiple Endocrine Neoplasia 2 in Denmark 1994-2014: A Nationwide Study. Thyroid 27 (2): 215-223, 2017.
- Kouvaraki MA, Shapiro SE, Perrier ND, et al.: RET proto-oncogene: a review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid 15 (6): 531-44, 2005.
- Kloos RT, Eng C, Evans DB, et al.: Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 19 (6): 565-612, 2009.
- Voss RK, Feng L, Lee JE, et al.: Medullary Thyroid Carcinoma in MEN2A: ATA Moderate- or High-Risk RET Mutations Do Not Predict Disease Aggressiveness. J Clin Endocrinol Metab 102 (8): 2807-2813, 2017.
- Mathiesen JS, Habra MA, Bassett JHD, et al.: Risk Profile of the RET A883F Germline Mutation: An International Collaborative Study. J Clin Endocrinol Metab 102 (6): 2069-2074, 2017.
- Machens A, Lorenz K, Weber F, et al.: Lymph node metastasis in hereditary medullary thyroid cancer is independent of the underlying RET germline mutation. Eur J Surg Oncol 47 (4): 920-923, 2021.
- Eng C, Smith DP, Mulligan LM, et al.: Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum Mol Genet 3 (2): 237-41, 1994.
- Hofstra RM, Landsvater RM, Ceccherini I, et al.: A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 367 (6461): 375-6, 1994.
- Carlson KM, Dou S, Chi D, et al.: Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci U S A 91 (4): 1579-83, 1994.
- Gimm O, Marsh DJ, Andrew SD, et al.: Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918 mutation. J Clin Endocrinol Metab 82 (11): 3902-4, 1997.
- Smith DP, Houghton C, Ponder BA: Germline mutation of RET codon 883 in two cases of de novo MEN 2B. Oncogene 15 (10): 1213-7, 1997.
- Eng C, Mulligan LM, Healey CS, et al.: Heterogeneous mutation of the RET proto-oncogene in subpopulations of medullary thyroid carcinoma. Cancer Res 56 (9): 2167-70, 1996.
- Cranston AN, Carniti C, Oakhill K, et al.: RET is constitutively activated by novel tandem mutations that alter the active site resulting in multiple endocrine neoplasia type 2B. Cancer Res 66 (20): 10179-87, 2006.
- Miyauchi A, Futami H, Hai N, et al.: Two germline missense mutations at codons 804 and 806 of the RET proto-oncogene in the same allele in a patient with multiple endocrine neoplasia type 2B without codon 918 mutation. Jpn J Cancer Res 90 (1): 1-5, 1999.
- Kameyama K, Okinaga H, Takami H: RET oncogene mutations in 75 cases of familial medullary thyroid carcinoma in Japan. Biomed Pharmacother 58 (6-7): 345-7, 2004 Jul-Aug.
- Iwashita T, Murakami H, Kurokawa K, et al.: A two-hit model for development of multiple endocrine neoplasia type 2B by RET mutations. Biochem Biophys Res Commun 268 (3): 804-8, 2000.
- Menko FH, van der Luijt RB, de Valk IA, et al.: Atypical MEN type 2B associated with two germline RET mutations on the same allele not involving codon 918. J Clin Endocrinol Metab 87 (1): 393-7, 2002.
- Mulligan LM, Eng C, Healey CS, et al.: Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat Genet 6 (1): 70-4, 1994.
- Seri M, Celli I, Betsos N, et al.: A Cys634Gly substitution of the RET proto-oncogene in a family with recurrence of multiple endocrine neoplasia type 2A and cutaneous lichen amyloidosis. Clin Genet 51 (2): 86-90, 1997.
- Rothberg AE, Raymond VM, Gruber SB, et al.: Familial medullary thyroid carcinoma associated with cutaneous lichen amyloidosis. Thyroid 19 (6): 651-5, 2009.
- Quayle FJ, Fialkowski EA, Benveniste R, et al.: Pheochromocytoma penetrance varies by RET mutation in MEN 2A. Surgery 142 (6): 800-5; discussion 805.e1, 2007.
- Bolino A, Schuffenecker I, Luo Y, et al.: RET mutations in exons 13 and 14 of FMTC patients. Oncogene 10 (12): 2415-9, 1995.
- Boccia LM, Green JS, Joyce C, et al.: Mutation of RET codon 768 is associated with the FMTC phenotype. Clin Genet 51 (2): 81-5, 1997.
- Lesueur F, Cebrian A, Cranston A, et al.: Germline homozygous mutations at codon 804 in the RET protooncogene in medullary thyroid carcinoma/multiple endocrine neoplasia type 2A patients. J Clin Endocrinol Metab 90 (6): 3454-7, 2005.
- Shannon KE, Gimm O, Hinze R: Germline V804M mutation in the RET protooncogene in 2 apparently sporadic cases of MTC presenting in the 7th decade of life. The Journal of Endocrine Genetics 1 (1): 39-46, 1999.
- Raue F, Frank-Raue K: Genotype-phenotype relationship in multiple endocrine neoplasia type 2. Implications for clinical management. Hormones (Athens) 8 (1): 23-8, 2009 Jan-Mar.
- Mulligan LM, Marsh DJ, Robinson BG, et al.: Genotype-phenotype correlation in multiple endocrine neoplasia type 2: report of the International RET Mutation Consortium. J Intern Med 238 (4): 343-6, 1995.
- Moers AM, Landsvater RM, Schaap C, et al.: Familial medullary thyroid carcinoma: not a distinct entity? Genotype-phenotype correlation in a large family. Am J Med 101 (6): 635-41, 1996.
- Niccoli-Sire P, Murat A, Rohmer V, et al.: Familial medullary thyroid carcinoma with noncysteine ret mutations: phenotype-genotype relationship in a large series of patients. J Clin Endocrinol Metab 86 (8): 3746-53, 2001.
- Machens A, Ukkat J, Brauckhoff M, et al.: Advances in the management of hereditary medullary thyroid cancer. J Intern Med 257 (1): 50-9, 2005.
- Mukherjee S, Zakalik D: RET codon 804 mutations in multiple endocrine neoplasia 2: genotype-phenotype correlations and implications in clinical management. Clin Genet 79 (1): 1-16, 2011.
- Xu JY, Grubbs EG, Waguespack SG, et al.: Medullary Thyroid Carcinoma Associated with Germline RETK666N Mutation. Thyroid 26 (12): 1744-1751, 2016.
- Erlic Z, Hoffmann MM, Sullivan M, et al.: Pathogenicity of DNA variants and double mutations in multiple endocrine neoplasia type 2 and von Hippel-Lindau syndrome. J Clin Endocrinol Metab 95 (1): 308-13, 2010.
- Toledo RA, Hatakana R, Lourenço DM, et al.: Comprehensive assessment of the disputed RET Y791F variant shows no association with medullary thyroid carcinoma susceptibility. Endocr Relat Cancer 22 (1): 65-76, 2015.
- Høxbroe Michaelsen S, Ornstrup MJ, Poulsen MM, et al.: Long-term follow-up of RET Y791F carriers in Denmark 1994-2017: A National Cohort Study. J Surg Oncol 119 (6): 687-693, 2019.
- Siqueira DR, Ceolin L, Ferreira CV, et al.: Role of RET genetic variants in MEN2-associated pheochromocytoma. Eur J Endocrinol 170 (6): 821-8, 2014.
- Ceolin L, Siqueira DR, Romitti M, et al.: Molecular basis of medullary thyroid carcinoma: the role of RET polymorphisms. Int J Mol Sci 13 (1): 221-39, 2012.
- Robledo M, Gil L, Pollán M, et al.: Polymorphisms G691S/S904S of RET as genetic modifiers of MEN 2A. Cancer Res 63 (8): 1814-7, 2003.
- Kaczmarek-Ryś M, Ziemnicka K, Pławski A, et al.: Modifying impact of RET gene haplotypes on medullary thyroid carcinoma clinical course. Endocr Relat Cancer 25 (4): 421-436, 2018.
- Margraf RL, Crockett DK, Krautscheid PM, et al.: Multiple endocrine neoplasia type 2 RET protooncogene database: repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Hum Mutat 30 (4): 548-56, 2009.
- Wells SA, Donis-Keller H: Current perspectives on the diagnosis and management of patients with multiple endocrine neoplasia type 2 syndromes. Endocrinol Metab Clin North Am 23 (1): 215-28, 1994.
- Gardet V, Gatta B, Simonnet G, et al.: Lessons from an unpleasant surprise: a biochemical strategy for the diagnosis of pheochromocytoma. J Hypertens 19 (6): 1029-35, 2001.
- Pacak K, Ilias I, Adams KT, et al.: Biochemical diagnosis, localization and management of pheochromocytoma: focus on multiple endocrine neoplasia type 2 in relation to other hereditary syndromes and sporadic forms of the tumour. J Intern Med 257 (1): 60-8, 2005.
- Raue F, Kraimps JL, Dralle H, et al.: Primary hyperparathyroidism in multiple endocrine neoplasia type 2A. J Intern Med 238 (4): 369-73, 1995.
- Milos IN, Frank-Raue K, Wohllk N, et al.: Age-related neoplastic risk profiles and penetrance estimations in multiple endocrine neoplasia type 2A caused by germ line RET Cys634Trp (TGC>TGG) mutation. Endocr Relat Cancer 15 (4): 1035-41, 2008.
- Marsh DJ, McDowall D, Hyland VJ, et al.: The identification of false positive responses to the pentagastrin stimulation test in RET mutation negative members of MEN 2A families. Clin Endocrinol (Oxf) 44 (2): 213-20, 1996.
- Elisei R, Romei C, Renzini G, et al.: The timing of total thyroidectomy in RET gene mutation carriers could be personalized and safely planned on the basis of serum calcitonin: 18 years experience at one single center. J Clin Endocrinol Metab 97 (2): 426-35, 2012.
- Rich TA, Feng L, Busaidy N, et al.: Prevalence by age and predictors of medullary thyroid cancer in patients with lower risk germline RET proto-oncogene mutations. Thyroid 24 (7): 1096-106, 2014.
- Qi XP, Zhao JQ, Du ZF, et al.: Prophylactic thyroidectomy for MEN 2-related medullary thyroid carcinoma based on predictive testing for RET proto-oncogene mutation and basal serum calcitonin in China. Eur J Surg Oncol 39 (9): 1007-12, 2013.
- Machens A, Lorenz K, Dralle H: Individualization of lymph node dissection in RET (rearranged during transfection) carriers at risk for medullary thyroid cancer: value of pretherapeutic calcitonin levels. Ann Surg 250 (2): 305-10, 2009.
- Castagna MG, Fugazzola L, Maino F, et al.: Reference range of serum calcitonin in pediatric population. J Clin Endocrinol Metab 100 (5): 1780-4, 2015.
- Kuhlen M, Frühwald MC, Dunstheimer DPA, et al.: Revisiting the genotype-phenotype correlation in children with medullary thyroid carcinoma: A report from the GPOH-MET registry. Pediatr Blood Cancer 67 (4): e28171, 2020.
- Schreinemakers JM, Vriens MR, Valk GD, et al.: Factors predicting outcome of total thyroidectomy in young patients with multiple endocrine neoplasia type 2: a nationwide long-term follow-up study. World J Surg 34 (4): 852-60, 2010.
- Skinner MA, Moley JA, Dilley WG, et al.: Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med 353 (11): 1105-13, 2005.
- Szinnai G, Meier C, Komminoth P, et al.: Review of multiple endocrine neoplasia type 2A in children: therapeutic results of early thyroidectomy and prognostic value of codon analysis. Pediatrics 111 (2): E132-9, 2003.
- Brauckhoff M, Machens A, Lorenz K, et al.: Surgical curability of medullary thyroid cancer in multiple endocrine neoplasia 2B: a changing perspective. Ann Surg 259 (4): 800-6, 2014.
- Ordóñez J, Pérez-Egido L, García-Casillas MA, et al.: Management and results of thyroidectomies in pediatric patients with MEN 2 syndrome. J Pediatr Surg 56 (11): 2058-2061, 2021.
- Franc S, Niccoli-Sire P, Cohen R, et al.: Complete surgical lymph node resection does not prevent authentic recurrences of medullary thyroid carcinoma. Clin Endocrinol (Oxf) 55 (3): 403-9, 2001.
- Zenaty D, Aigrain Y, Peuchmaur M, et al.: Medullary thyroid carcinoma identified within the first year of life in children with hereditary multiple endocrine neoplasia type 2A (codon 634) and 2B. Eur J Endocrinol 160 (5): 807-13, 2009.
- Hansen HS, Torring H, Godballe C, et al.: Is thyroidectomy necessary in RET mutations carriers of the familial medullary thyroid carcinoma syndrome? Cancer 89 (4): 863-7, 2000.
- Moley JF, Skinner M, Gillanders WE, et al.: Management of the Parathyroid Glands During Preventive Thyroidectomy in Patients With Multiple Endocrine Neoplasia Type 2. Ann Surg 262 (4): 641-6, 2015.
- Machens A, Schneyer U, Holzhausen HJ, et al.: Prospects of remission in medullary thyroid carcinoma according to basal calcitonin level. J Clin Endocrinol Metab 90 (4): 2029-34, 2005.
- Spanheimer PM, Ganly I, Chou J, et al.: Long-Term Oncologic Outcomes After Curative Resection of Familial Medullary Thyroid Carcinoma. Ann Surg Oncol 26 (13): 4423-4429, 2019.
- Razvi S, Hostalek U: Therapeutic challenges in the application of serum thyroid stimulating hormone testing in the management of patients with hypothyroidism on replacement thyroid hormone therapy: a review. Curr Med Res Opin 35 (7): 1215-1220, 2019.
- Sawin CT, Geller A, Hershman JM, et al.: The aging thyroid. The use of thyroid hormone in older persons. JAMA 261 (18): 2653-5, 1989.
- Baloch Z, Carayon P, Conte-Devolx B, et al.: Laboratory medicine practice guidelines. Laboratory support for the diagnosis and monitoring of thyroid disease. Thyroid 13 (1): 3-126, 2003.
- Seib CD, Harari A, Conte FA, et al.: Utility of serum thyroglobulin measurements after prophylactic thyroidectomy in patients with hereditary medullary thyroid cancer. Surgery 156 (2): 394-8, 2014.
- Samaan NA, Schultz PN, Hickey RC: Medullary thyroid carcinoma: prognosis of familial versus nonfamilial disease and the role of radiotherapy. Horm Metab Res Suppl 21: 21-5, 1989.
- Scherübl H, Raue F, Ziegler R: Combination chemotherapy of advanced medullary and differentiated thyroid cancer. Phase II study. J Cancer Res Clin Oncol 116 (1): 21-3, 1990.
- Subbiah V, Hu MI, Wirth LJ, et al.: Pralsetinib for patients with advanced or metastatic RET-altered thyroid cancer (ARROW): a multi-cohort, open-label, registrational, phase 1/2 study. Lancet Diabetes Endocrinol 9 (8): 491-501, 2021.
- Wirth LJ, Sherman E, Robinson B, et al.: Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N Engl J Med 383 (9): 825-835, 2020.
- Wells SA, Robinson BG, Gagel RF, et al.: Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol 30 (2): 134-41, 2012.
- Fox E, Widemann BC, Chuk MK, et al.: Vandetanib in children and adolescents with multiple endocrine neoplasia type 2B associated medullary thyroid carcinoma. Clin Cancer Res 19 (15): 4239-48, 2013.
- Kraft IL, Akshintala S, Zhu Y, et al.: Outcomes of Children and Adolescents with Advanced Hereditary Medullary Thyroid Carcinoma Treated with Vandetanib. Clin Cancer Res 24 (4): 753-765, 2018.
- Elisei R, Schlumberger MJ, Müller SP, et al.: Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol 31 (29): 3639-46, 2013.
- Schlumberger M, Elisei R, Müller S, et al.: Overall survival analysis of EXAM, a phase III trial of cabozantinib in patients with radiographically progressive medullary thyroid carcinoma. Ann Oncol 28 (11): 2813-2819, 2017.
- Sherman SI, Clary DO, Elisei R, et al.: Correlative analyses of RET and RAS mutations in a phase 3 trial of cabozantinib in patients with progressive, metastatic medullary thyroid cancer. Cancer 122 (24): 3856-3864, 2016.
- Carlomagno F, Guida T, Anaganti S, et al.: Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 23 (36): 6056-63, 2004.
- Subbiah V, Velcheti V, Tuch BB, et al.: Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol 29 (8): 1869-1876, 2018.
- Drilon A, Hu ZI, Lai GGY, et al.: Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol 15 (3): 151-167, 2018.
- Redaelli S, Plaza-Menacho I, Mologni L: Novel targeted therapeutics for MEN2. Endocr Relat Cancer 25 (2): T53-T68, 2018.
- Mulligan LM: Progress and potential impact of RET kinase targeting in cancer. Expert Rev Proteomics 13 (7): 631-3, 2016.
- Lenders JW, Duh QY, Eisenhofer G, et al.: Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 99 (6): 1915-42, 2014.
- Castinetti F, Qi XP, Walz MK, et al.: Outcomes of adrenal-sparing surgery or total adrenalectomy in phaeochromocytoma associated with multiple endocrine neoplasia type 2: an international retrospective population-based study. Lancet Oncol 15 (6): 648-55, 2014.
- Machens A, Lorenz K, Weber F, et al.: Recurrent ipsilateral pheochromocytoma in carriers of RET p.Cys634 missense mutations. Endocrine 77 (1): 160-167, 2022.
- Lairmore TC, Ball DW, Baylin SB, et al.: Management of pheochromocytomas in patients with multiple endocrine neoplasia type 2 syndromes. Ann Surg 217 (6): 595-601; discussion 601-3, 1993.
- Inabnet WB, Caragliano P, Pertsemlidis D: Pheochromocytoma: inherited associations, bilaterality, and cortex preservation. Surgery 128 (6): 1007-11;discussion 1011-2, 2000.
- Scholten A, Valk GD, Ulfman D, et al.: Unilateral subtotal adrenalectomy for pheochromocytoma in multiple endocrine neoplasia type 2 patients: a feasible surgical strategy. Ann Surg 254 (6): 1022-7, 2011.
- Grubbs EG, Rich TA, Ng C, et al.: Long-term outcomes of surgical treatment for hereditary pheochromocytoma. J Am Coll Surg 216 (2): 280-9, 2013.
- Walz MK, Alesina PF, Wenger FA, et al.: Posterior retroperitoneoscopic adrenalectomy--results of 560 procedures in 520 patients. Surgery 140 (6): 943-8; discussion 948-50, 2006.
- Walz MK, Alesina PF, Wenger FA, et al.: Laparoscopic and retroperitoneoscopic treatment of pheochromocytomas and retroperitoneal paragangliomas: results of 161 tumors in 126 patients. World J Surg 30 (5): 899-908, 2006.
- Perrier ND, Kennamer DL, Bao R, et al.: Posterior retroperitoneoscopic adrenalectomy: preferred technique for removal of benign tumors and isolated metastases. Ann Surg 248 (4): 666-74, 2008.
- Behrman SW, Bahr MH, Dickson PV, et al.: The microbiology of secondary and postoperative pancreatic infections: implications for antimicrobial management. Arch Surg 146 (5): 613-9, 2011.
- Evans DB, Perrier ND: On "Posterior retroperitoneoscopic adrenalectomy--results of 560 procedures in 520 patients". Surgery 140 (6): 951-2, 2006.
- Dickson PV, Jimenez C, Chisholm GB, et al.: Posterior retroperitoneoscopic adrenalectomy: a contemporary American experience. J Am Coll Surg 212 (4): 659-65; discussion 665-7, 2011.
- Cabalag MS, Mann GB, Gorelik A, et al.: Posterior retroperitoneoscopic adrenalectomy: outcomes and lessons learned from initial 50 cases. ANZ J Surg 85 (6): 478-82, 2015.
- Norton JA, Brennan MF, Wells SA Jr: Surgical Management of Hyperparathyroidism. In: Bilezikian JP, Marcus R, Levine MA: The Parathyroids: Basic and Clinical Concepts. Raven Press, 1994, pp 531-551.
- Scholten A, Schreinemakers JM, Pieterman CR, et al.: Evolution of surgical treatment of primary hyperparathyroidism in patients with multiple endocrine neoplasia type 2A. Endocr Pract 17 (1): 7-15, 2011 Jan-Feb.
- Machens A, Dralle H: Advances in risk-oriented surgery for multiple endocrine neoplasia type 2. Endocr Relat Cancer 25 (2): T41-T52, 2018.
- Khan MI, Waguespack SG, Hu MI: Medical management of postsurgical hypoparathyroidism. Endocr Pract 17 (Suppl 1): 18-25, 2011 Mar-Apr.
- Stålberg P, Carling T: Familial parathyroid tumors: diagnosis and management. World J Surg 33 (11): 2234-43, 2009.
- Peacock M, Bilezikian JP, Klassen PS, et al.: Cinacalcet hydrochloride maintains long-term normocalcemia in patients with primary hyperparathyroidism. J Clin Endocrinol Metab 90 (1): 135-41, 2005.
- Schuffenecker I, Ginet N, Goldgar D, et al.: Prevalence and parental origin of de novo RET mutations in multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma. Le Groupe d'Etude des Tumeurs a Calcitonine. Am J Hum Genet 60 (1): 233-7, 1997.
- Norum RA, Lafreniere RG, O'Neal LW, et al.: Linkage of the multiple endocrine neoplasia type 2B gene (MEN2B) to chromosome 10 markers linked to MEN2A. Genomics 8 (2): 313-7, 1990.
- Carlson KM, Bracamontes J, Jackson CE, et al.: Parent-of-origin effects in multiple endocrine neoplasia type 2B. Am J Hum Genet 55 (6): 1076-82, 1994.
- Kitamura Y, Scavarda N, Wells SA, et al.: Two maternally derived missense mutations in the tyrosine kinase domain of the RET protooncogene in a patient with de novo MEN 2B. Hum Mol Genet 4 (10): 1987-8, 1995.
- Rich TA, Liu M, Etzel CJ, et al.: Comparison of attitudes regarding preimplantation genetic diagnosis among patients with hereditary cancer syndromes. Fam Cancer 13 (2): 291-9, 2014.
- Freyer G, Ligneau B, Schlumberger M, et al.: Quality of life in patients at risk of medullary thyroid carcinoma and followed by a comprehensive medical network: trends for future evaluations. Ann Oncol 12 (10): 1461-5, 2001.
- Freyer G, Dazord A, Schlumberger M, et al.: Psychosocial impact of genetic testing in familial medullary-thyroid carcinoma: a multicentric pilot-evaluation. Ann Oncol 10 (1): 87-95, 1999.
- Grosfeld FJ, Lips CJ, Ten Kroode HF, et al.: Psychosocial consequences of DNA analysis for MEN type 2. Oncology (Huntingt) 10 (2): 141-6; discussion 146, 152, 157, 1996.
- Johnston LB, Chew SL, Trainer PJ, et al.: Screening children at risk of developing inherited endocrine neoplasia syndromes. Clin Endocrinol (Oxf) 52 (2): 127-36, 2000.
- MacDonald DJ, Lessick M: Hereditary cancers in children and ethical and psychosocial implications. J Pediatr Nurs 15 (4): 217-25, 2000.
- Grosfeld FJ, Lips CJ, Beemer FA, et al.: Psychological risks of genetically testing children for a hereditary cancer syndrome. Patient Educ Couns 32 (1-2): 63-7, 1997 Sep-Oct.
- Giarelli E: Multiple endocrine neoplasia type 2a (MEN2a): a call for psycho-social research. Psychooncology 11 (1): 59-73, 2002 Jan-Feb.
- Grosfeld FJ, Lips CJ, Beemer FA, et al.: Distress in MEN 2 family members and partners prior to DNA test disclosure. Multiple endocrine neoplasia type 2. Am J Med Genet 91 (1): 1-7, 2000.
- Grosfeld FJ, Beemer FA, Lips CJ, et al.: Parents' responses to disclosure of genetic test results of their children. Am J Med Genet 94 (4): 316-23, 2000.
- Giarelli E: Bringing threat to the fore: participating in lifelong surveillance for genetic risk of cancer. Oncol Nurs Forum 30 (6): 945-55, 2003 Nov-Dec.
- Schultz PN: Providing information to patients with a rare cancer: using Internet discussion forums to address the needs of patients with medullary thyroid carcinoma. Clin J Oncol Nurs 6 (4): 219-22, 2002 Jul-Aug.